

Abstract
Background The increased permeability of the blood-brain barrier (BBB) is a critical contributor to the high mortality following ischaemic stroke. However, the mechanisms regulating BBB integrity remain poorly understood. Leucine-rich repeat-containing 8A (LRRC8A) is a chloride channel critical for cellular volume homeostasis and plays a key role in regulating neuronal injury during ischaemia. However, its impact on BBB function is currently unclear.
Methods A transient middle cerebral artery occlusion model was established to investigate the impact of LRRC8A on BBB integrity. Laser speckle contrast imaging was used to monitor cortical blood flow. Primary mouse and human brain microvascular endothelial cells (m/hBMVECs) were subjected to oxygen-glucose deprivation (OGD) and re-oxygenation for varying durations. Patch-clamp recordings were performed to measure volume-regulated chloride currents. Immunostaining was conducted to evaluate protein expression. Cell permeability was evaluated with transwell assay.
Results LRRC8A deletion in endothelial cells ameliorates the infarct area and mitigates BBB leakage. Ischaemia dramatically upregulates the expression of LRRC8A in endothelial cells, concurrently downregulating tight junction proteins. OGD exposure augments the VRCC current mediated by LRRC8A in BMVECs. In contrast, inhibiting LRRC8A promotes the expression of ZO-1 and VE-cadherin, thereby preserving the integrity of endothelial cells. With-no-lysine kinase 1 (WNK1) inhibition contributes to LRRC8A-induced BBB damage post-ischaemic stroke. Eupatorin, a newly identified LRRC8A inhibitor, exerts neuroprotective effects against ischaemic stroke.
Conclusions LRRC8A in BMVECs plays a pivotal role in modulating BBB integrity, a process regulated by WNK1. As an LRRC8A inhibitor, Eupatorin holds the potential for ischaemic stroke therapy.
WHAT IS ALREADY KNOWN ON THIS TOPIC
Blood-brain barrier (BBB) damage is a primary cause of secondary injury following ischaemic stroke.
Leucine-rich repeat-containing 8A (LRRC8A) contributes to the neuronal excitotoxicity in the context of ischaemic stroke.
WHAT THIS STUDY ADDS
LRRC8A in endothelial cells contributes to the aberrant BBB integrity in ischaemic stroke.
With-no-lysine kinase 1 mediates LRRC8A-dependent regulation of the BBB.
Eupatorin, as an LRRC8A inhibitor, holds potential for ischaemic stroke therapy.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
LRRC8A in the cerebral vascular endothelial cells represents a promising target for modulating BBB integrity.
Introduction
The integrity of the blood-brain barrier (BBB) is crucial for the maintenance of brain microenvironment homeostasis, composed of endothelial cells, glial cells, pericytes and extracellular matrix.1 A remarkable pathological feature of ischaemic stroke is the changes in the permeability of BBB, which further triggers brain oedema and poor prognosis.2 The tight junction between brain microvascular endothelial cells (BMVECs) plays a key role in the ‘barrier’, which can maintain the low permeability of BBB and protect the central nervous system from the potentially toxic substances in the blood. The tight junction and adherens junction protein complexes, including ZO-1, occludin and VE-cadherin, form the basis of the tight junction structures in endothelial cells. The abnormality of tight junctions can lead to the disruption of the BBB and the development of related brain diseases.3
Ion channel regulation is a central mechanism underlying BBB homeostasis. A large body of ion channels, including KCa3.1, TREK and TMEM16A, have been shown to modulate the tight junction property, mostly through the modulation of endothelial [Ca2+]i.4–6 Volume-regulated chloride channel (VRCC) is crucial for maintaining cell volume, with leucine-rich repeat-containing 8A (LRRC8A) serving as the core molecular component, potentially in a cell-type-dependent manner.7 Modulation of LRRC8A function holds potential for brain protection. Conditional knockout of the LRRC8A in hippocampal neurons has been shown to benefit brain function during ischaemia-reperfusion.8 Additionally, LRRC8A channel activity in astrocytes may differentially regulate neuronal activity in conditions such as ischaemia and drug addiction.9 10 Moreover, LRRC8A in endothelial cells is crucial for maintaining vascular function and LRRC8A conditional knockout facilitated the formation of moderate hypertension.11 Ischaemia-induced BBB disruption is often accompanied by endothelial swelling.12 13 However, whether this volume-related regulation of BBB integrity involves LRRC8A has yet to be investigated.
With-no-lysine kinase 1 (WNK1) is a widely distributed kinase family in mammals, named for the absence of a catalytic lysine in its subdomain II.14 WNK1 serves as an intracellular Cl− sensor and plays a role in modulating endothelial barrier function.15 Additionally, WNK1 functions as a molecular crowding sensor and becomes active in response to cell shrinkage triggered by a hypertonic extracellular solution.16 It regulates transmembrane ion movement by regulating Cl− transporters NKCC1 and KCC activity through phosphorylation of SPS/Ste20-related proline-alanine-rich kinase (STK39) and oxidative stress responsive 1 (OXSR1), thereby influencing cell volume increase.16 A previous study has demonstrated that LRRC8A overexpression promotes cell proliferation and vascular remodelling via the WNK1/PI3K/AKT signalling pathway.17 However, whether LRRC8A-mediated modulation of WNK1 activity regulates endothelial tight junctions and BBB integrity remains elusive.
In the present study, we aim to test the hypothesis that cerebral ischaemia potentiates the activity of endothelial LRRC8A-dependent VRCC, which contribute to BBB disruption and ischaemic brain injury through WNK1 inhibition. Furthermore, we have identified Eupatorin as a specific inhibitor of the LRRC8A channel, demonstrating neuroprotective effects in a mouse model of cerebral ischaemia. Our study provides novel insights into the role of endothelial LRRC8A in ischaemic brain injury and its potential clinical implications.
Materials and methods
Animals
Mice were housed in groups of five per cage in a controlled animal facility maintained at a temperature of 23°C±2°C and a humidity range of 30%–50%. The mice followed a 12-hour light/dark cycle and had ad libitum access to food and water. Mice were randomly assigned to experimental groups using a blinded allocation protocol. The LRRC8A flox allele was generated by Biocytogen Pharmaceuticals (Beijing) based on the CRISPR/Cas9 approach. The 5′loxP is inserted into intron 2 and 3′loxP is inserted into intron 3. LRRC8A flox allele was crossed with the endothelial Tie-2Cre, all fully in the C57Bl/6 background, to generate LRRC8A flox/flox; Tie-2 Cre mice.
Full experimental methods are shown in online supplemental materials.
Supplementary data
Results
Conditional knockout of LRRC8A in endothelial cells ameliorates brain injury post-ischaemic stroke
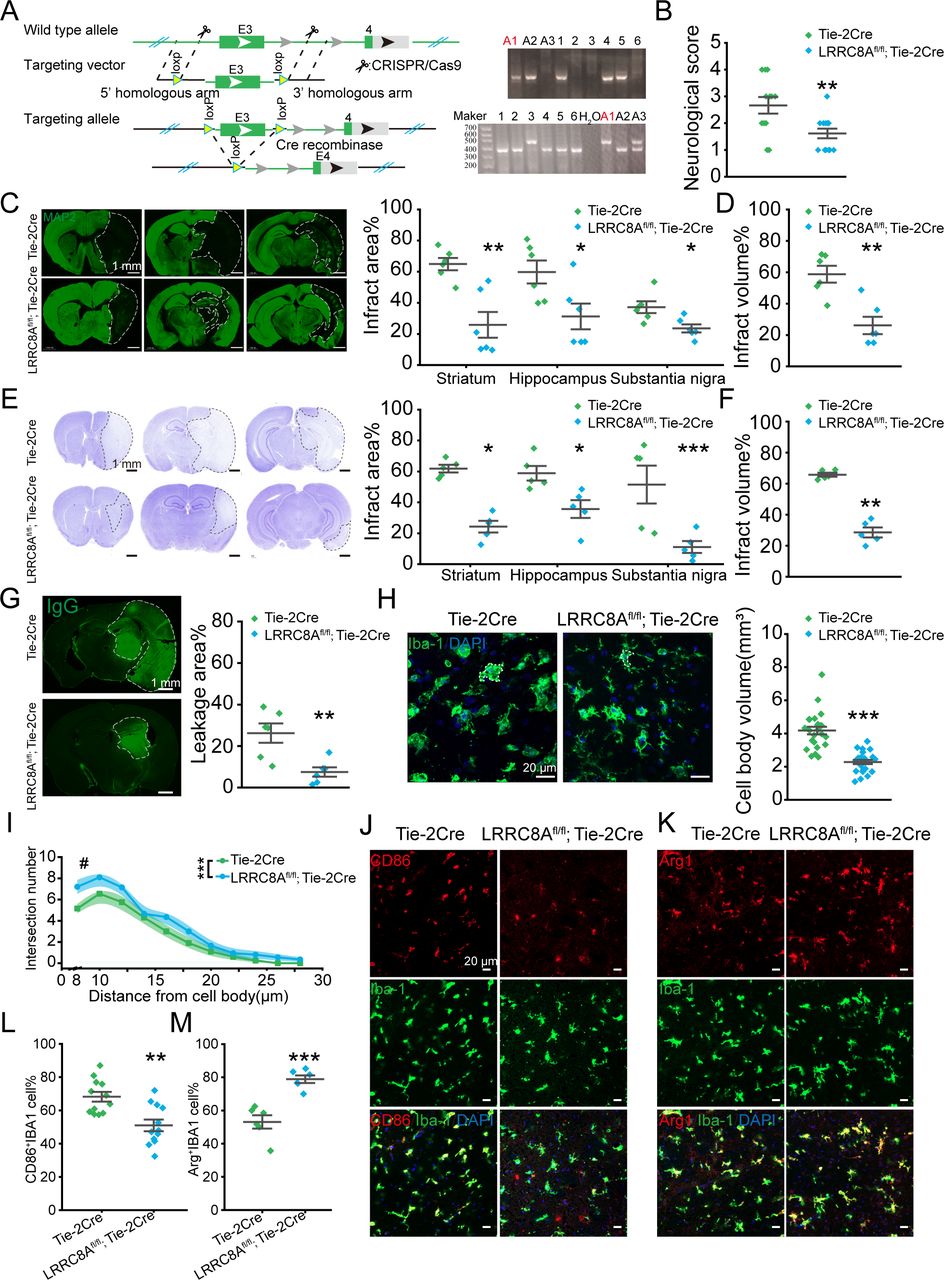
To investigate the role of LRRC8A in BMVECs in BBB regulation following ischaemic stroke, we generated mice with endothelial cell-specific knockout of LRRC8A, designated as LRRC8Afl/fl; Tie-2Cre mice as illustrated in figure 1A. Validation of gene knockout using fluorescence staining and PCR was presented in online supplemental figure S1. Mice were subjected to middle cerebral artery occlusion (MCAO) for 90 min, followed by reperfusion. Changes in the cerebral blood flow were shown in online supplemental figure S2. Results indicated that endothelial cell-specific knockout of the LRRC8A significantly improved neurological scores 24 hours after reperfusion (figure 1B).

Conditional knockout (cKO) of LRRC8A in endothelial cells could reduce brain injury post-ischaemic stroke. (A) Illustration of LRRC8A knockout with the CRISPR/Cas9 strategy. The right panel was the DNA genotype identification and the mice marked with ‘A1’ was the LRRC8Afl/fl; Tie-2Cre one. The genotype of LRRC8Afl/fl; Tie-2Cre was designated as the LRRC8A cKO mice and Tie-2Cre was the little-mate negative control. (B) The neurological scores 24 hours after ischaemia-reperfusion. N=13 mice in Tie-2Cre group, n=12 mice in LRRC8Afl/fl; Tie-2Cre group, **p<0.01, two-sample t-test. (C) Left panel: representative images of MAP2 immunostaining 24 hours after MCAO surgery. The white dotted lines indicated the borders of the infarct zones. The position of the brain slices from left to right was Bregma 0.86 mm, −1.34 mm and −2.80 mm. Right panel: quantification of the infarct areas across coronal sections. N=6 mice in Tie-2Cre group, n=6 mice in LRRC8Afl/fl; Tie-2Cre group, *p<0.05, **p<0.01, two-way ANOVA. (D) Quantification of total infarct volume. N=6 mice in the Tie-2Cre group, n=6 mice in the LRRC8Afl/fl; Tie-2Cre group, **p<0.01, two-sample t-test. (E) Left panel: representative images of Nissl staining 24 hours after MCAO. The white dotted lines indicated the borders of the infarct zones. The position of the brain slices from left to right was Bregma 0.86 mm, −1.34 mm and −2.80 mm. Right panel: quantification of the infarct areas across coronal sections. N=5 mice in Tie-2Cre group, n=5 mice in LRRC8Afl/fl; Tie-2Cre group, *p<0.05, ***p<0.001, two-way ANOVA. (F) Quantification of total infarct volume. N=5 mice in Tie-2Cre group, n=5 mice in LRRC8Afl/fl; Tie-2Cre group, **p<0.01, two-sample t-test. (G) Representative images demonstrated the extravasation of IgG into the brain parenchyma. The white dotted lines indicated the area of IgG leakage. The right panel was the quantitative analysis of IgG leakage area. N=6 mice in each group, **p<0.01, two-sample t-test. (H) Representative images of microglia in the Tie-2Cre and LRRC8Afl/fl; Tie-2Cre group. The right panel was the quantitative analysis of microglial soma volume. N=24 cells from 6 mice in each group. ***P<0.001, two-sample t-test. (I) Sholl analysis of microglia. N=20 cells from 5 mice in each group. #P<0.05, ***p<0.001, two-way repeated ANOVA. (J, K) Representative images of CD86, Arg1 and IBA1 co-staining in the thalamus in peri-infarct regions from Tie-2 Cre and LRRC8Afl/fl; Tie-2Cre mice. (L) Quantification of co-staining of CD86 and IBA1. N=12 views from 6 mice in each group. **P<0.01, two-sample t-test. (M) Quantification of co-staining of Arg1 and IBA1. N=6 views from 6 mice in each group. ***P<0.001, two-sample t-test. ANOVA, analysis of variance; LRRC8A, leucine-rich repeat-containing 8A; MAP2, microtubule-associated protein 2.
Microtubule-associated protein 2 (MAP2) immunostaining was performed to evaluate the ischaemic area according to previous studies.18–20 As illustrated in figure 1C,D, the infarct area indicated with MAP2-negative staining was significantly reduced with endothelial cell-specific knockout of LRRC8A. This finding was corroborated by Nissl staining, which showed a dramatic attenuation in the infarct area (figure 1E,F). In addition, the BBB leakage triggered by ischaemia-reperfusion (figure 1G) was significantly reduced, as indicated by IgG-positive staining. To be noted, we compared the vascular structure and the coverage of astrocytes and pericytes between LRRC8Afl/fl; Tie-2Cre and Tie-2Cre mice strains. The findings revealed no significant differences between the two groups (online supplemental figure S3).
Microglia residing within the thalamus in the peri-infarct region were evaluated based on the cell morphology, revealing a reduction in the cell body volume (figure 1H) and an increase in the protrusions (figure 1I) in the LRRC8Afl/fl; Tie-2Cre mice group. Furthermore, a notable difference was observed in the expression of CD86 and Arg1 in microglia between the two groups. Specifically, the endothelial cell-specific knockout of LRRC8A mice exhibited a significant elevation in the proportion of Arg1+ microglia and a marked decrease in the proportion of CD86+ microglia 24 hours after reperfusion (figure 1J–M). Based on previous research,21–23 the alterations in microglial morphology and molecular expression demonstrated a reduction in the inflammatory response of microglia following ischaemic-reperfusion and an increase in the population of microglia with tissue repair functions, further supporting the role of LRRC8A downregulation in endothelial cells in ameliorating ischaemic brain injury.
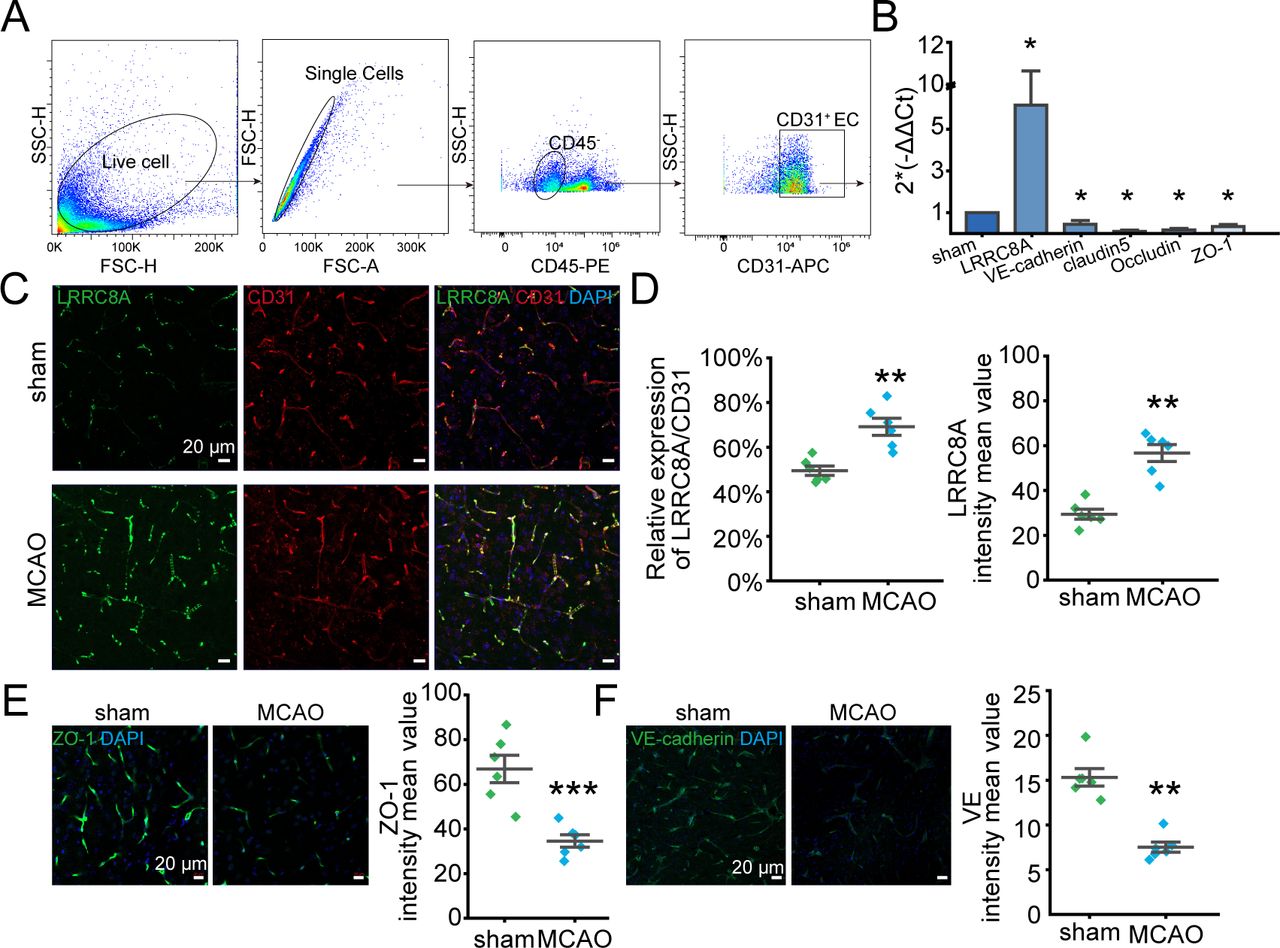
Expression of LRRC8A in endothelial cells is elevated post-ischaemic stroke
To further elucidate the mechanism underlying LRRC8A’s regulation of the BBB, we first assessed the expression of LRRC8A in vascular endothelial cells after ischaemia-reperfusion. As illustrated in figure 2A, brain vascular endothelial cells were isolated by fluorescence-activated cell sorting, and mRNA expression levels of LRRC8A along with tight junction and adherens junction markers were quantified. As shown in figure 2B, the mRNA level of LRRC8A was dramatically elevated in the MCAO group. Correspondingly, the mRNAs including claudin5, occludin, VE-cadherin and ZO-1 were significantly decreased 24 hours after reperfusion. Consistently, the expression level of LRRC8A in CD31 positive cells (endothelial cells) significantly increased 24 hours after ischaemia-reperfusion (figure 2C,D). Additionally, both ZO-1 and VE-cadherin protein expression levels were notably downregulated following cerebral ischaemia and reperfusion (figure 2E,F).

LRRC8A expression was augmented in endothelial cells post-ischaemic stroke. Mice underwent MCAO surgery and sham operation 24 hours after reperfusion, LRRC8A expression in endothelial cells was tested. CD31 was used as the marker of endothelial cells. (A) CD45−CD31+ was used as the strategy of brain endothelial cell sorting. (B) mRNA levels of LRRC8A, VE-cadherin, claudin5, occludin and ZO-1 in sorted endothelial cells in sham and MCAO groups. Data were normalised to the sham group. Three mice were used for endothelial cell sorting each time and data represent three independent experiments. *P<0.05, two-sample t-test. (C) Representative images showed the co-expression of LRRC8A and CD31 in the sham and MCAO groups. (D) Analysis of fluorescence density. N=6 mice in each group. **P<0.01, two-sample t-test. (E, F) Left panel: representative images of ZO-1 (E) and VE-cadherin (F) in sham and MCAO groups. Right panel: quantification of ZO-1 and VE-cadherin expression. N=6 mice in each group. **P<0.01, two-sample t-test. LRRC8A, leucine-rich repeat-containing 8A; MCAO, middle cerebral artery occlusion.
LRRC8A channel function is augmented with oxygen-glucose deprivation exposure
Oxygen-glucose deprivation (OGD) induced by ischaemic stroke will increase the volume of endothelial cells, which can trigger the VRCC current in hippocampal CA1 pyramidal neurons.8 12 13 VRCC function in endothelial cells was further evaluated with OGD exposure to simulate ischaemic stroke conditions in vitro (figure 3A). Hypertonic pipette solution (420 mOsm) induced VRCC currents were measured in both human brain microvascular endothelial cells (hBMVECs) and primary mouse brain microvascular endothelial cells (mBMVECs). Figure 3C–E and G–I illustrated the typical slow activation of VRCC current at −60 mV following cell swelling, with outwardly rectifying characteristics and was inhibited by a selective VRCC blocker DCPIB. OGD exposure induced a substantial increase in VRCC current density in both hBMVECs and mBMVECs (figure 3F,J). To prove that LRRC8A is the major contributor of VRCC current observed in hBMVECs and mBMVECs, VRCC currents were also observed in these cells on specific siRNA-mediated knockdown of LRRC8A (figure 3B). As shown in figure 3K–R, the VRCC currents in these LRRC8A-siRNA transfected cells were significantly downregulated in comparison with the control cells, suggesting LRRC8A was responsible for encoding VRCC current in both hBMVECs and mBMVECs.

OGD augmented the VRCC current in both hBMVECs and mBMVECs that was mediated by LRRC8A. (A, B) Illustration of the experiment designs. (C) The hBMVECs were held at −60 mV and the currents were recorded using a hypertonic (420 mOsm) pipette solution. The representative traces of VRCC currents were inhibited by DCPIB (10 µM) at −60 mV preconditioning with OGD or under control conditions. (D, E) The representative current traces induced by step (D) or ramp (E) protocols were shown when the VRCC current in panel C reached the maximum at −60 mV preconditioning with OGD or under control conditions. (F) Summary data for the hBMVECs current densities recorded at −60 mV. N=12 cells in each group. *P<0.05, compared with hBMVECs control. (G) VRCC current was recorded with mBMVECs at −60 mV. (H, I) The representative current traces induced by step (H) or ramp (I) protocols were shown when the VRCC current in panel G reached the maximum at −60 mV. (J) Summary data for the mBMVECs current densities recorded at −60 mV. N=12 cells in each group. *P<0.05, compared with the mBMVECs group. (K) The representative VRCC current traces in the hBMVECs transfected with LRRC8A siRNA or scramble. (L, M) The representative current traces induced by step (L) or ramp (M) protocols were shown when the VRCC current in panel K reached the maximum at −60 mV. (N) Summary data for the hBMVECs VRCC current densities recorded at −60 mV transfected with LRRC8A siRNA or scramble. N=10 cells in each group. *P<0.05, compared with hBMVECs group. (O) The representative VRCC current traces in the mBMVECs transfected with LRRC8A siRNA or scramble. (P, Q) The representative current traces induced by step (P) or ramp (Q) protocols were shown when the VRCC current in panel O reached the maximum at −60 mV. (R) Summary data for the mBMVECs VRCC current densities recorded at −60 mV transfected with LRRC8A siRNA or scramble. N=10 cells in each group. *P<0.05, compared with mBMVECs group. All statistical analyses were performed using two-sample t-tests. hBMVEC, human brain microvascular endothelial cell; LRRC8A, leucine-rich repeat-containing 8A; mBMVEC, mouse brain microvascular endothelial cell; OGD, oxygen-glucose deprivation; VRCC, volume-regulated chloride channel.
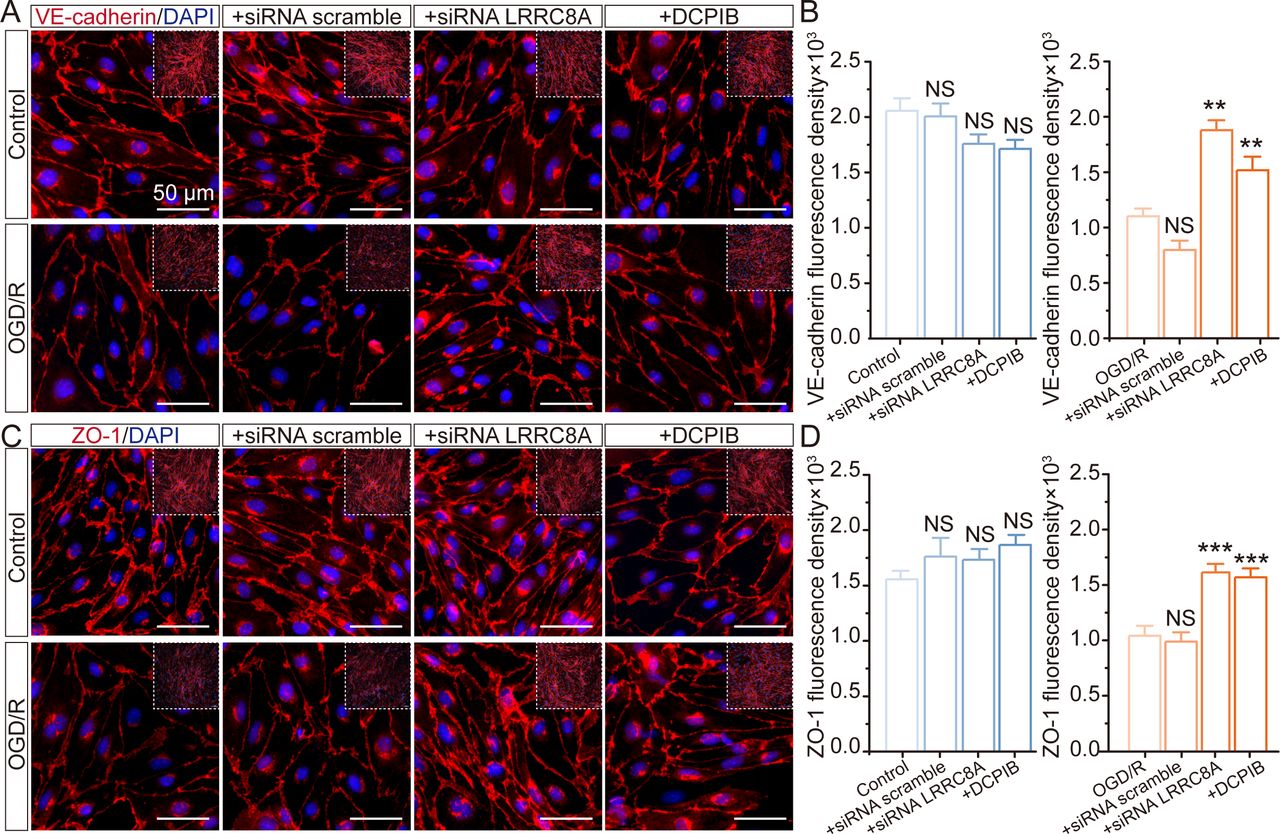
Deletion of LRRC8A improves the tight junction between endothelial cells
The tight junction between vascular endothelial cells plays a pivotal role in preserving the integrity of the BBB. To investigate the role of LRRC8A in BBB regulation, we examined the effects of LRRC8A gene knockdown on the overall expression of tight junction and adherens junction proteins, including VE-cadherin and ZO-1, in cultured mBMVECs. As depicted in figure 4A,B, cerebral vascular endothelial cells normally formed dense junctions like paving stones, marked with VE-cadherin, while OGD/re-oxygenation (OGD/R) significantly induced a reduction in the overall expression of VE-cadherin within mBMVECs. The application of LRRC8A siRNA remarkably mitigated this effect, with no discernible impact on expression under standard culture conditions. We also evaluated the impact of the VRCC blocker DCPIB on VE-cadherin in mBMVECs, yielding results consistent with siRNA performance. Similarly, OGD/R also led to a substantial reduction in the overall expression of ZO-1 in mBMVECs as shown in figure 4C,D. The application of LRRC8A siRNA or DCPIB could significantly restore ZO-1 expression but did not affect its levels under normal culture conditions.

Effect of LRRC8A modulation on ZO-1 and VE-cadherin expression in mBMVECs. mBMVECs were isolated and cultured for the BBB model in vitro. Four days later, a confluent monolayer with a tight junction between endothelial cells was produced. Then LRRC8A siRNA or scramble control was transfected. 48 hours later, cells were administrated with OGD exposure for 12 hours and followed by re-oxygenation (R) for 24 hours. Additionally, mBMVECs were pre-incubated with DCPIB (10 µM) for 30 min before OGD/R exposure. (A) and (C) Representative images (zoomed-in images of the inner part) showed the ZO-1 and VE-cadherin expression in different groups. (B) and (D) Quantification of ZO-1 and VE-cadherin expression in both the plasma membrane and cytoplasm from the inner part with the NIS-Elements AR Analysis software (V.5.01.00). Data represented the fluorescence intensity per unit area and were derived from four independent experiments. ***P<0.001, compared with the first column. **P<0.05, ***p<0.001, compared with the first column, one-way analysis of variance. BBB, blood-brain barrier; LRRC8A, leucine-rich repeat-containing 8A; mBMVEC, mouse brain microvascular endothelial cell; OGD, oxygen-glucose deprivation.
Deletion of LRRC8A mitigates OGD/R-induced BBB permeability
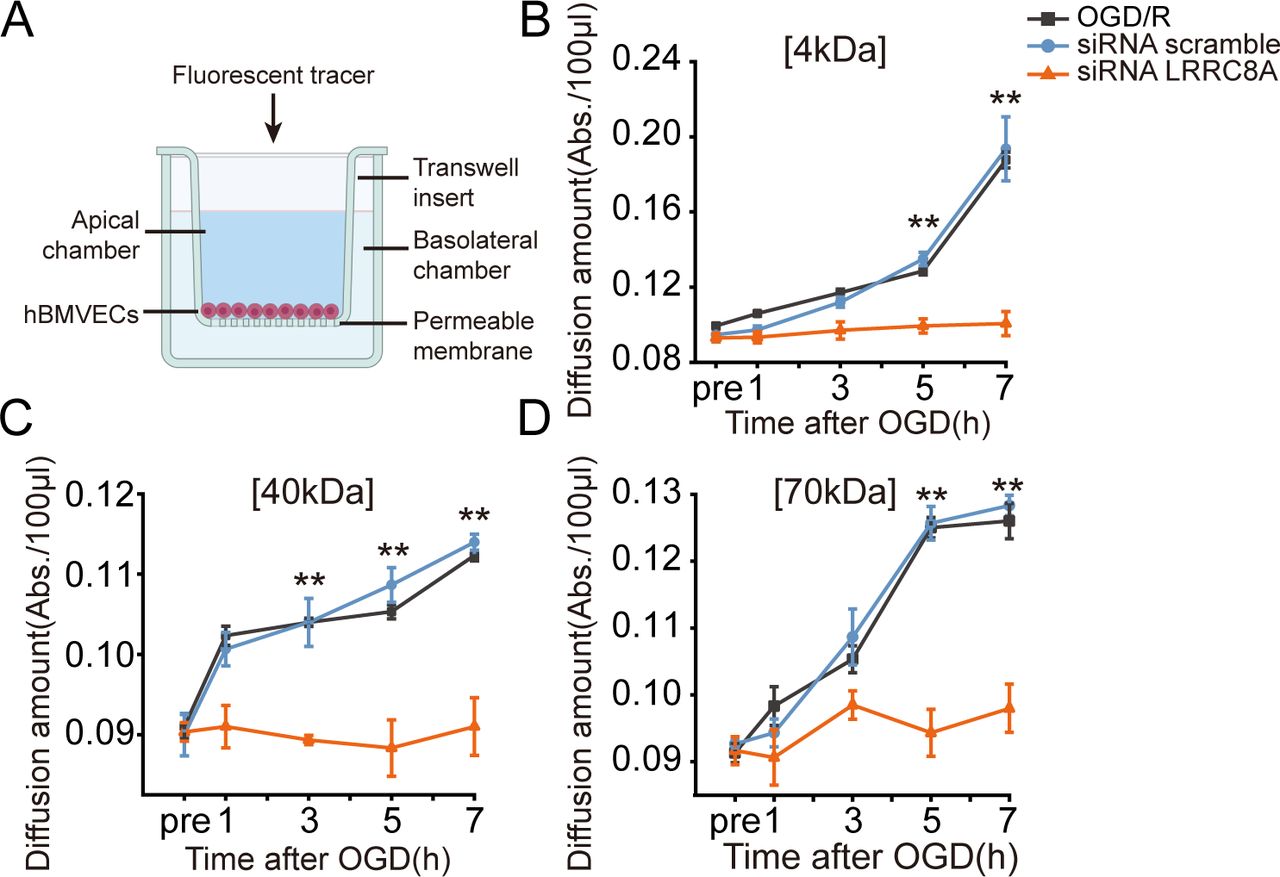
Deprivation of oxygen and glucose can also lead to the increased permeability of microvascular endothelial cells. To further validate the role of LRRC8A in this process, hBMVECs were seeded on transwell inserts, and fluorescent tracers with different molecular weights (4 kDa, 40 kDa and 70 kDa FITC-dextran) were added into cells within the inserts to test the diffusion of each tracer in the lower chamber of the transwell at different time points post-OGD, which served as an indicator of cell permeability.
Data shown in figure 5 revealed that as the duration of re-oxygenation increased, hBMVECs permeability to FITC-dextrans of three different molecular weights increased sequentially. Notably, at the 7-hour post-OGD, the permeability of 4 kDa FITC-dextran was the most pronounced. Importantly, knockdown of the LRRC8A gene with specific siRNA led to a significant reduction in the permeability of all three tracers, compared with the scramble group, suggesting that LRRC8A gene knockdown could ameliorate BBB permeability induced by OGD/R. To be noticed, 12 hours of OGD followed by up to 7 hours of re-oxygenation did not cause significant cell death in cultured hBMVECs (online supplemental figure S4).

LRRC8A knockout restored BBB integrity after OGD in vitro. (A) Schematic of the in vitro endothelial cell permeability assay. hBMVECs were seeded on the top insert and grown for 4 days to form a confluent monolayer. Then, hBMVECs were transfected with LRRC8A siRNA or scramble control for 48 hours, followed by OGD exposure for 12 hours. Cell permeability was evaluated at 1-, 3-, 5- and 7-hour post-OGD by the luminal to abluminal diffusion amount of three fluorescent tracers with different molecular weights. (B–D) Diffusion amounts of 4 kDa (B), 40 kDa (C) and 70 kDa (D) FITC-dextran at 1-, 3-, 5- and 7-hour post-OGD. Data represent six independent experiments. **P<0.01, compared with the scramble group, two-way analysis of variance. BBB, blood-brain barrier; hBMVEC, human brain microvascular endothelial cell; LRRC8A, leucine-rich repeat-containing 8A; OGD, oxygen-glucose deprivation.
WNK1 mediates the regulatory effect of LRRC8A on the expression of tight junction proteins
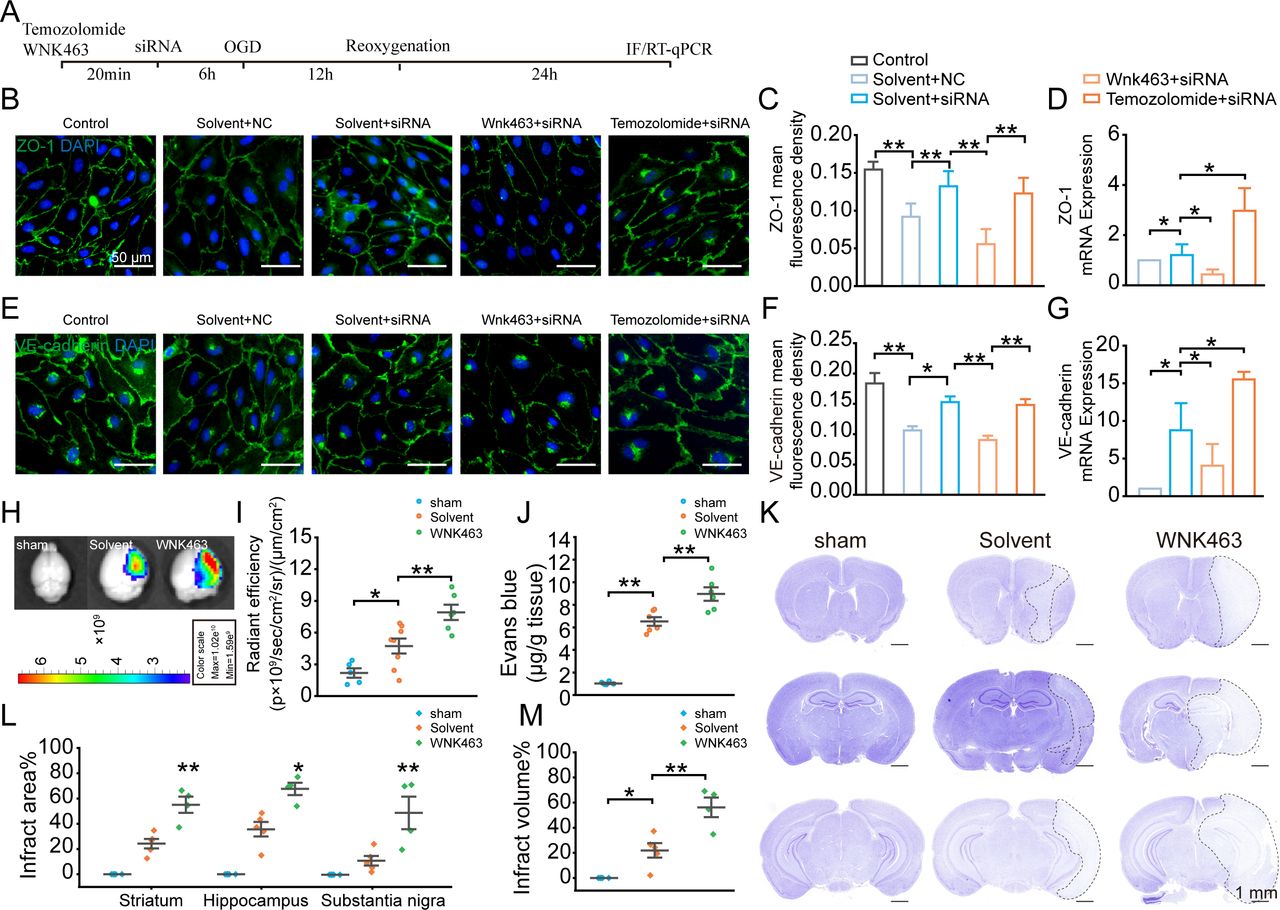
WNK1 is essential for BBB function, and most importantly, WNK1 has been reported to be a chloride sensor.15 24 Chloride could stabilise the inactive conformation of WNK1 and prevent kinase activation. Hence, we next investigated whether WNK1 participates in the regulatory process of LRRC8A on the expression of tight junction proteins. The experimental workflow is depicted in figure 6A. As shown in figure 6B,C and E,F, LRRC8A gene knockdown significantly increased the expression levels of both ZO-1 and VE-cadherin. However, preconditioning incubation of WNK1 inhibitor WNK463 significantly reduced both ZO-1 and VE-cadherin expression, while WNK1 activator Temozolomide increased the expression of both ZO-1 and VE-cadherin. The effect of the WNK1 regulator on the mRNA levels of ZO-1 and VE-cadherin was consistent with its impact on protein expression, as depicted in figure 6D,G.

WNK1-mediated LRRC8A-induced modulation of tight junctions. (A) Illustration of the experimental workflow. mBMVECs were incubated with 10 µM WNK463 (WNK1 inhibitor), 10 µM Temozolomide (WNK1 agonist) or solvent for 20 min, followed by transfection with siRNA LRRC8A or siRNA scramble. 48 hours later, the cells were subjected to OGD, followed by 24 hours of re-oxygenation. RNA was then extracted from the cells, or the cells were subjected to immunofluorescence staining to detect the expression levels of ZO-1 and VE-cadherin in each group. (B) Representative images of ZO-1 expression under different conditions. (C) Quantification of ZO-1 fluorescence density in both the plasma membrane and cytoplasm with ImageJ software (V.1.52a). Data represented the fluorescence intensity per unit area and were derived from three independent experiments. (D) Quantification of ZO-1 mRNA level. Data represented three independent experiments. *P<0.05, **p<0.01, one-way ANOVA. (E) Representative images of VE-cadherin expression under different conditions. (F) Quantification of VE-cadherin fluorescence density in both the plasma membrane and cytoplasm with ImageJ software (V.1.52a). Data represented the fluorescence intensity per unit area. (G) Quantification of VE-cadherin mRNA level. *P<0.05, **p<0.01, one-way ANOVA. (H–J) Endothelial cell-specific knockout of LRRC8A mice underwent sham and MCAO operation. Solvent and WNK463 were administrated before surgery. Representative images (H) and statistical analysis (I) demonstrated the extravasation of EB into the cortex. N=5 mice in sham group, n=8 mice in solvent group, n=6 mice in WNK463 group, *p<0.05, **p<0.01, one-way ANOVA. (J) Quantification of EB leakage with tissue homogenisation. N=6 mice in each group, **p<0.01, one-way ANOVA. (K) Representative Nissl-stained coronal sections 24 hours post-MCAO in endothelial cell-specific LRRC8A knockout mice. The white dotted lines indicated borders of the infarct zones. Sections were arranged from rostral to caudal at Bregma coordinates: 0.86 mm, −1.34 mm and −2.80 mm. (L) Quantification of the infarct area across coronal sections. N=5 mice in sham group, n=5 mice in solvent group, n=4 mice in WNK463 group, *p<0.05, **p<0.01, two-way ANOVA. (M) Quantification of total infarct volume. N=5 mice in sham group, n=5 mice in solvent group, n=4 mice in WNK463 group, *p<0.05, **p<0.01, one-way ANOVA. ANOVA, analysis of variance; BBB, blood-brain barrier; mBMVEC, mouse brain microvascular endothelial cell; LRRC8A, leucine-rich repeat-containing 8A; MCAO, middle cerebral artery occlusion; OGD, oxygen-glucose deprivation; WNK1, with-no-lysine kinase 1.
WNK463 was further used in vivo to detect its role in LRRC8A deletion-mediated BBB protection. We used LRRC8A endothelial cell-specific knockout mice to investigate the role of WNK1. These mice underwent either a sham procedure or MCAO surgery. As shown in figure 6H,I, administration of 10 mg/kg WNK463 through intravenous injection prior to the MCAO model could significantly increase the leakage of Evans blue (EB). EB leakage detected with the tissue homogenisation method confirmed this finding further (figure 6J). In addition, WNK463 could increase the infarct volume, compared with the vehicle group (figure 6K–M), suggesting the vital function of WNK1 in LRRC8A-mediated BBB damage and brain injury.
LRRC8A channel inhibitor, Eupatorin contributes to the protection of BBB integrity and reduction of infarct area post-ischaemic stroke
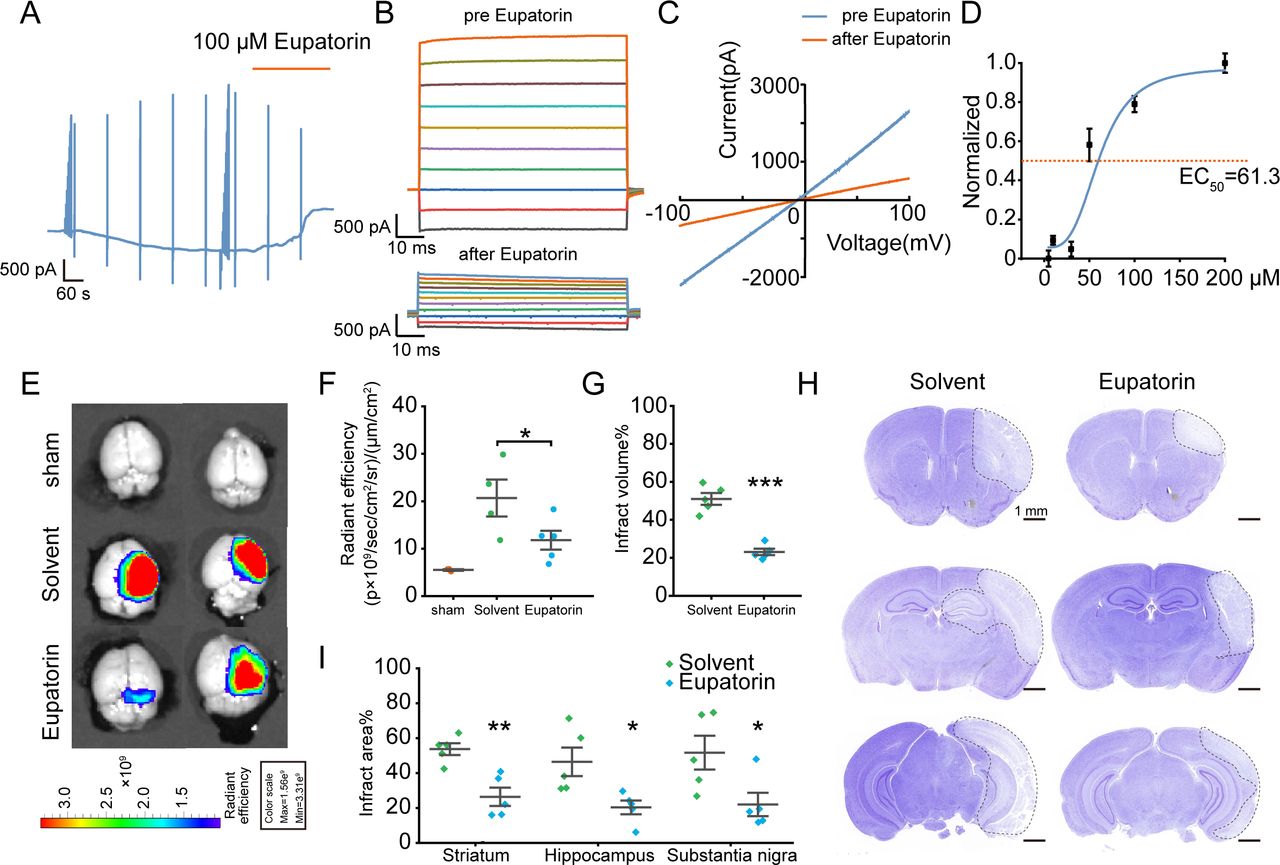
Our preliminary research demonstrated that the flavonoid compound Eupatorin could effectively block the LRRC8A channel in HEK293A cells.25 We first investigated the impact of Eupatorin on LRRC8A currents in hBMVECs and found that Eupatorin could significantly inhibit LRRC8A currents in a dose-dependent manner (figure 7A–C). The EC50 was 61.3 µM (figure 7D). Furthermore, the IVISSpectrum CT Imaging System was used to observe the effect of Eupatorin on the BBB leakage following cerebral ischaemia with wild-type mice. 24 hours after ischaemia-reperfusion, mice were administered EB via tail vein injection, followed by cardiac perfusion and brain extraction for imaging. As shown in figure 7E,F, Eupatorin markedly decreased the EB content in the brain parenchyma. Nissl staining further revealed that Eupatorin could significantly minimise the infarct volume (figure 7G–I).

Effect of Eupatorin on LRRC8A and brain injury post-ischaemic stroke. (A) hBMVECs were continuously held at −60 mV and stimulated with 1 s voltage ramps from −100 to +100 mV applied with a 60 s interval, 100 µM Eupatorin was applied when the current reached the maximum. (B) Effect of Eupatorin on the current–voltage (I/V) relationship, with voltage steps ranging from −100 to +100 mV in 20 mV increments. (C) The representative current traces induced by ramp protocols were shown before and Eupatorin application. (D) Dose–response curve of Eupatorin and LRRC8A current inhibition. N=6 cells for analysis. (E, F) Representative images (E) and statistical analysis (F) demonstrated the extravasation of EB into the cortex pretreated with Eupatorin or vehicle in wild-type mice. N=3 mice in sham group, n=4 mice in solvent group, n=5 in Eupatorin group, *p<0.05, one-way ANOVA. (G) Quantification of total infarct volume. N=5 mice in each group, ***p<0.001, two-sample t-test. (H) Representative images of Nissl staining. The white dotted lines indicated the borders of the infarct zones. Sections were arranged from rostral to caudal at Bregma coordinates: 0.86 mm, −1.34 mm and −2.80 mm. (I) Quantification of the infarct area across coronal sections. N=5 mice in each group, *p<0.05, **p<0.01, two-way ANOVA. ANOVA, analysis of variance; hBMVEC, human brain microvascular endothelial cell; LRRC8A, leucine-rich repeat-containing 8A.
Discussion
The present study investigated the role of LRRC8A in BMVECs in BBB damage after ischaemia-reperfusion. Ischaemia-induced upregulation of LRRC8A in endothelial cells leads to disruption of tight junctions and subsequent BBB leakage. Endothelial cell-specific knockout of LRRC8A will benefit the BBB restoration and reduce the brain infarction area.
Given that ischaemia can induce cell swelling,26 VRCC activation might be a primary factor in the disruption of the BBB post-ischaemic stroke. The role of LRRC8A as the molecular basis for VRCC remains somewhat controversial.7 Our study demonstrates the significant contribution of LRRC8A to VRCC currents in BMVECs and confirms that OGD will trigger the functional augmentation of LRRC8A in endothelial cells.
We further investigated the effect of hypotonic solution on ZO-1 and VE-cadherin in microvascular endothelial cells. Consistent with previous research,27 hypotonic stress significantly reduced the expression levels of these proteins, as demonstrated in online supplemental figure S5. However, the cell swelling will return to normal over time due to the regulated volume decrease28 or exposure to the isotonic solution, suggesting that tight junction protein expression is weakly correlated with cellular volume regulation but strongly associated with LRRC8A channel activation and its downstream signalling pathways.
Further investigations revealed that inhibiting the LRRC8A function could maintain the low permeability of endothelial cells post OGD/R. Using fluorescent tracers of three different molecular weights, we observed that endothelial cell permeability to the 4 kDa indicator was higher than the 70 kDa indicator. This finding aligns with previous studies and supports the reliability of our in vitro BBB model.19 Additionally, with increased re-oxygenation time, cell permeability to all three fluorescent indicators significantly increased, suggesting aggravated reperfusion injury.
Following OGD and subsequent re-oxygenation, endothelial cell volume partially recovered, which was LRRC8A-dependent (online supplemental figure S6). However, re-oxygenation or reperfusion triggered secondary oxidative stress responses, causing further cellular damage.29 This process might no longer depend on LRRC8A-mediated volume regulation. Some studies have indicated that LRRC8A can regulate nitric oxide synthase production in a volume-independent manner, ultimately promoting the generation of inflammatory factors.30 Our research suggests that ischaemic injury can trigger endothelial cell swelling, activate LRRC8A channels and mediate BBB damage. The additional damage to the BBB after reperfusion could be an additive effect arising from the functional regulation of LRRC8A.
We further focus on the signalling pathway involved in LRRC8A-mediated BBB modulation. WNK1 has been reported to be activated in response to low intracellular chloride concentrations.15 24 Chloride could stabilise the inactive conformation of WNK1 and prevent kinase activation. As a chloride channel, LRRC8A is activated in response to cell swelling, leading to the alterations of intracellular chloride concentration that potentially modulate WNK1 activity. As previously demonstrated, WNK1 influences VE-cadherin expression in a TGF-β dependent manner.31 WNK1 could enhance the stability of TGF-β dependent signalling factors, including SMAD2/3, RhoA, and ALK1. WNK463, as a WNK1 inhibitor, could lead to a decreased VE-cadherin staining pattern, consistent with a need for WNK1 to promote adherens junctions.32
In addition, the effect of WNK1 on tight junctions is also related to changes in intracellular calcium. Research indicated that inhibition of the cystic fibrosis transmembrane conductance regulator, a chloride channel, could suppress WNK1 activation, elevate intracellular calcium levels and result in barrier leakage in pulmonary microvascular endothelial cells. Conversely, activation of WNK1 could enhance barrier function by inhibiting the rise in intracellular calcium concentration.33 During cerebral ischaemia, the calcium levels in vascular endothelial cells significantly increased, leading to the disruption of tight junctions via the calmodulin kinase pathway.34
Our results demonstrated that blocking the WNK1 function could attenuate the BBB protective effects induced by LRRC8A inhibition. However, it remains unclear whether this will involve the calcium/calmodulin signalling. Our previous research uncovered that LRRC8A gene knockout significantly suppressed the increase of intracellular calcium induced by fetal bovine serum, suggesting the role of LRRC8A in modulating calcium changes.35 Moving forward, we will continue to investigate the role of calcium signalling in the regulation of tight junctions in BMVECs. Additionally, further research will aim to identify the signalling molecules through which WNK1 influences intracellular calcium levels, including STK39 and OXSR1.
Another compelling avenue of discussion pertains to the response of microglia to endothelial cell-specific knockout of LRRC8A post-ischaemia. Microglia are well known for their crucial role in regulating brain injury and angiogenesis.36 It is intriguing to explore how the functional regulation of LRRC8A in endothelial cells influences the state of microglia, providing a novel perspective for studying the interaction between the vascular and immune systems. Studies have shown that LRRC8A plays a significant role in the release of neurotransmitters such as glutamate and GABA, as well as amino acids like taurine.37 38 In future studies, we will further investigate the mechanisms through which LRRC8A in cerebral endothelial cells influences microglia state, thereby providing new theoretical insights into brain injury caused by cerebral ischaemia.
Eupatorin has been demonstrated to modulate vascular relaxation and suppress tumour progression.39 40 The present study indicated that Eupatorin was an effective inhibitor of LRRC8A and suggested the role of Eupatorin in reducing BBB leakage and infarct areas, providing a potential therapeutic strategy for brain protection by targeting LRRC8A and advancing Eupatorin as a candidate drug for ischaemic stroke treatment.
In summary, we showed that LRRC8A was significantly upregulated in BMVECs post-ischaemic stroke, and endothelial cell-specific knockout of LRRC8A could alleviate ischaemic brain injury and BBB leakage. Furthermore, LRRC8A modulated the overall expression of tight junction proteins and endothelial permeability through WNK1 regulation, thus promoting BBB damage. These findings reveal LRRC8A as a promising target for suppressing BBB leakage and brain injury after ischaemic stroke, and LRRC8A inhibitor, Eupatorin, could be a candidate for ischaemic stroke therapy.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
All animal care and experimental procedures complied with the guidelines and were approved by the Animal Ethics Committee.
Footnotes
YT and YW are joint first authors.
YT and YW contributed equally.
Contributors HuiranZ and HailinZ designed the project. HuiranZ, YT, YW, CL, YZhao, YZhang and YK conducted behaviour tests and biological experiments. YT, YW, CL, XZ, YZhao, BW and ZW established mice model and performed immunostaining. HuaxingZ and XD provided methods and technical supports. HuiranZ and HailinZ conceived and supervised the project. HuiranZ drafted the manuscript. HuiranZ and HailinZ revised and finalised the manuscript writing. HuiranZ and HailinZ, as guarantors, funded this study and were responsible for the overall content. All authors read and agreed to the final manuscript. YT and YW contributed equally to this paper.
Funding This study was funded by National Natural Science Foundation of China (No.81871075, No.82071533, No.U21A20359, No.8187087), Science Fund for Creative Research Groups of Natural Science Foundation (H2020206474), Basic Research Fund for Provincial Universities grant (JCYJ2021010), Central Guidance on Local Science and Technology Development grant (226Z7703G).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
References




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}