

Abstract
Background Neuroinflammation participates in the pathogenesis of subarachnoid haemorrhage (SAH); however, no effective treatments exist. MicroRNAs regulate several aspects of neuronal dysfunction. In a previous study, we found that exosomal miR-486-3p is involved in the pathophysiology of SAH. Targeted delivery of miR-486-3p without blood-brain barrier (BBB) restriction to alleviate SAH is a promising neuroinflammation approach.
Methods In this study, we modified exosomes (Exo) to form an RVG-miR-486-3p-Exo (Exo/miR) to achieve targeted delivery of miR-486-3p to the brain. Neurological scores, brain water content, BBB damage, flow cytometry and FJC staining were used to determine the effect of miR-486-3p on SAH. Western blot analysis, ELISA and RT-qPCR were used to measure relevant protein and mRNA levels. Immunofluorescence staining and laser confocal detection were used to measure the expression of mitochondria, lysosomes and autophagosomes, and transmission electron microscopy was used to observe the level of mitophagy in the brain tissue of mice after SAH.
Results Tail vein injection of Exo/miR improved targeting of miR-486-3p to the brains of SAH mice. The injection reduced levels of neuroinflammation-related factors by changing the phenotype switching of microglia, inhibiting the expression of sirtuin 2 (SIRT2) and enhancing mitophagy. miR-486-3p treatment alleviated neurobehavioral disorders, brain oedema, BBB damage and neurodegeneration. Further research found that the mechanism was achieved by regulating the acetylation level of peroxisome proliferator-activated receptor γ coactivator l alpha (PGC-1α) after SIRT2 enters the nucleus.
Conclusion Exo/miR treatment attenuates neuroinflammation after SAH by inhibiting SIRT2 expression and stimulating mitophagy, suggesting potential clinical applications.
WHAT IS ALREADY KNOWN ON THIS TOPIC
Subarachnoid hemorrhage is a serious stroke disease, and the subsequent neuroinflammation seriously affects the patient’s prognosis. Due to the limitation of the blood-brain barrier, there is currently a lack of ideal methods to deliver drugs directly to brain tissue to alleviate neuroinflammation after subarachnoid hemorrhage (SAH).
WHAT THIS STUDY ADDS
Our study first confirmed that RVG-Lamp2b-Exo improve targeting of miR-486-3p to the brain after SAH, and Exo/miR alleviate neuroinflammation by microglial polarisation and promoting mitophagy, which was achieved by inhibiting SIRT2.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our study demonstrated that Exo/miR treatment attenuates neuroinflammation after SAH by inhibiting SIRT2 expression and stimulating mitophagy.
Introduction
Subarachnoid hemorrhage (SAH) is a lethal hemorrhagic cerebrovascular disease primarily caused by the rupture of intracranial aneurysms. It is one of the most devastating cerebrovascular diseases and is the second leading cause of death among stroke individuals. Approximately 30% of the survivors suffer permanent disabilities.1–3 Early brain injury (EBI) critically affects SAH outcomes. EBI involves several physiological disorders, among which, neuroinflammation is essential4–6—reducing the neuroinflammation of EBI after SAH is critical for improving outcomes. Many treatments include medications and gene therapies.7 8 MicroRNAs (miRNAs) are emerging gene therapies; they are a group of non-coding RNAs comprising 17–24 nucleotides, which regulate the expression of several target genes at the post-transcriptional level.9 Studies showed that miRNAs in the brain are significantly altered after SAH.10 11 Our previous studies reported the relationship between the levels of miRNAs and SAH outcomes.12–14 These reports suggested that miRNAs may enhance neurogenesis, angiogenesis and neuroplasticity. However, treatment is challenging due to the blood-brain barrier (BBB).15 Effective treatments without BBB limitations are urgently needed.
Exosomes are lipid bilayer membrane-enclosed nanoscale particles released from various cell types, acting as intercellular messengers by transferring proteins and nucleotides to neighbouring or distant cells and can freely pass through the BBB.16 17 Exos are emerging treatments in immunotherapy, regenerative medicine, drug delivery and other fields due to their low immunogenicity, good biodegradability, low toxicity, potent ability to protect endogenous bioactive components and the ability to pass through the BBB freely.18 19 However, natural exosomes lack targeting ability and accumulate rapidly in the liver and spleen after systemic administration instead of targeting specific tissues.20 Recent studies showed that targeted delivery exosomes could be achieved by expressing particular ligands on the membrane surface of Exo through gene modification.21 Alvarez-Erviti et al22 modified Exos of dendritic cells with acetylcholine receptor-specific rabies virus glycoprotein (RVG) peptides, enabling Exos to carry medications across the BBB. RVGs were engineered onto the membrane surface of exosomes via the fusion protein lysosome-associated membrane glycoprotein 2b (Lamp2b) to achieve neuron-specific targeting. Therefore, the specific targeted therapy can be achieved with the modified Exos.
The current study is based on the hypothesis that engineered Exos carrying miR-486-3p can be delivered to the brain and bind to SIRT2 to modulate neuroinflammatory damage in SAH mice. Further research showed that miR-486-3p delivered by RVG-Exo could alleviate inflammatory damage by regulating mitophagy through SIRT2, which may become an innovative strategy for treating SAH.
Material and methods
Patient and public involvement
No patients involved.
Ethics and animals
Study participants were recruited from the Department of Neurosurgery, the Nanjing Drum Tower Hospital, the affiliated hospital of Nanjing University Medical School. The study was performed following the Declaration of Helsinki. All patients were approved by the Ethics Committee of the Nanjing Drum Tower Hospital (2021–313–07), and all mice were performed according to the guidelines of the National Institutes of Health on the care and use of animals. Healthy adult male C57BL/6J mice (8–10 weeks, 18‒22 g) were purchased from the Animal Centre of Nanjing University Model Animal Research Centre, China. The mice were housed in a laboratory environment with temperature-controlled and humidity-controlled animal quarters, a 12-hour light/dark cycle and free access to water and food.
Results
RVG-EV-mediated miR-486-3p alleviates the neuroinflammation by microglial polarization
First, we successfully constructed and verified the delivery of engineering Exos (online supplemental figure S3), and then, we explored the role of Exo/miR in SAH mice. We determined whether Exo could be delivered to brain tissue. Immunofluorescence staining was used to stain the brain slices of SAH mice with Iba1 (microglia) and PKH26 (Exo) with DAPI and found that Exo-labelled miR-486-3p targeted microglia (figure 1), indicating that intravenously injected Exo can be taken up by microglia.
Supplementary data

Exo/miR alleviates neuroinflammation and improves neurological functions by microglial polarisation. A Exo were targeted deliver to brain successfully and labelled in microglia. B–E Immunofluorescence staining and quantitative analysis of Exo distributed in CD86 (M1) and CD206 (M2) of Sham, SAH, Exo, Exo/Scr and Exo/miR group. F–K qPCR and ELISA quantitative analysis of the level of neuroinflammation factors (IL-6, IL-1β, TNF-α) in each group at 24 hours after SAH. L Neurological score. M Evan’s blue and N brain water content in each group at 24 hours after SAH. O–P FJC staining and cell apoptosis in the cortex with quantification. ***p<0.001, ****p<0.0001. Scale bar: 20 μm.
To explore the role of Exo/miR in microglia, we divided the mice into Sham, SAH (24 hours), RVG-Exo (Exo), RVG-scrambled miRNA-Exo (Exo/Scr) and RVG-miR-486-3p-Exo (Exo/miR) groups, and then, we used CD86 (M1), CD206 (M2) and Iba1 (microglia) for immunofluorescence staining of brain tissues in each group. The results showed that CD86 expression was more significant in the SAH group than the Sham group, but there was no evident change in the Exo and Exo/Scr groups; however, levels were significantly lower in the Exo/miR group (figure 1B,D) (p<0.05). By contrast, CD206 expression was significantly lower in the SAH group and more significant in the Exo/miR group (figure 1C,E) (p<0.05). These findings suggest that many M1 phenotype microglia appear, accompanied by a large amount of neuroinflammation after SAH. When Exo/miR was injected, the neuroinflammation was inhibited, and M1 microglia were replaced by M2 microglia.
To determine the role of Exo/miR in neuroinflammation, we measured expression levels of inflammatory factors such as IL-1β, IL-6 and TNF-α using RT-qPCR and ELISA. These results were consistent with the previous description (figure 1F-K),
To explore the impact of Exo/miR on neurological function, we measured postoperative neurological function scores, brain tissue oedema and BBB damage in SAH mice. We found that neurological function scores in the SAH group were lower than in the Sham group; after treatment with Exo/miR, neurological function scores moderately recovered (figure 1L). When treated with Exo/miR, brain oedema and Evans blue levels were significantly lower than the Sham group (figure 1M,N) (p<0.05). These findings suggest that Exo/miR improves neurological function scores of mice after SAH and reduces brain oedema and BBB damage.
Then, we tested whether Exo/miR can rescue neuronal degeneration and apoptosis. FJC staining and FCM showed that neuronal degeneration and apoptosis in the SAH group were higher than in the Sham group, while the Exo group and Exo/Scr group showed no evident changes; when treated with Exo/miR, neuronal degeneration and apoptosis were significantly reduced (figure 1O,P) (p<0.05).
Exo/miR alleviates neuroinflammation by promoting mitophagy
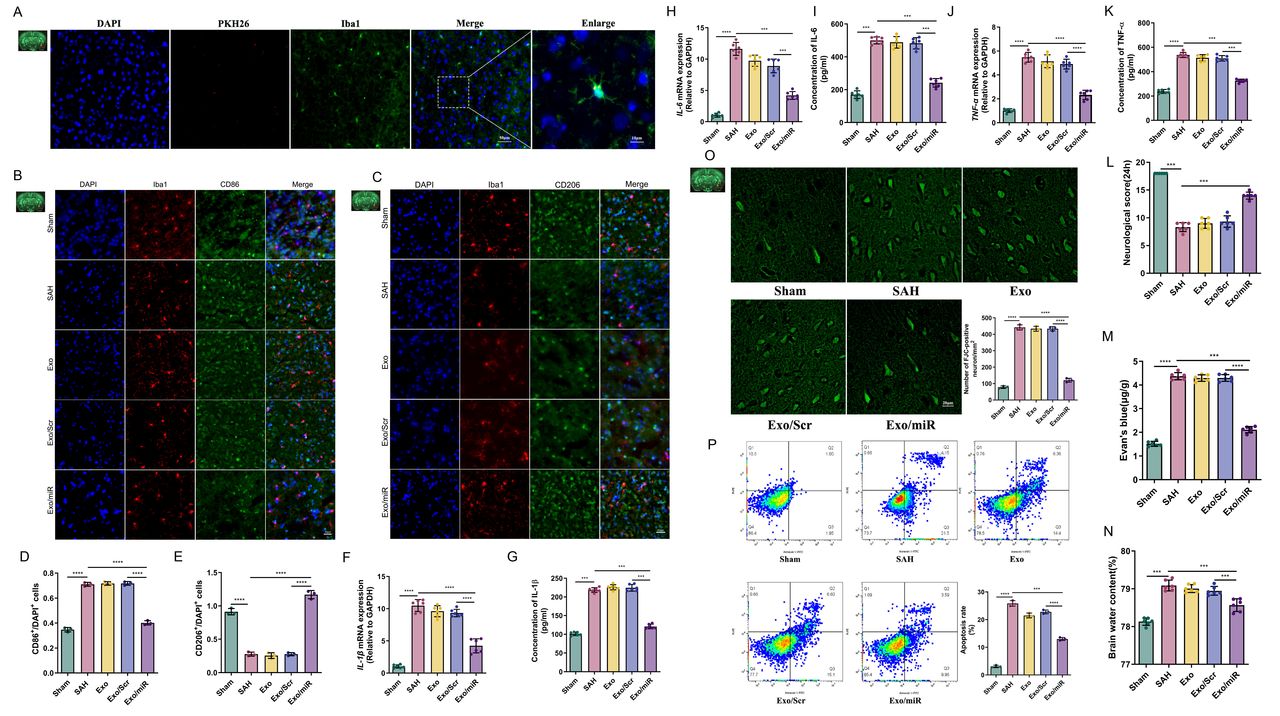
Mitophagy is essential in neuroinflammation after SAH.23 We designed the following experiments to explore whether Exo/miR could affect mitophagy. Parkin and Pink1 are critical for mitochondrial damage, and their expression levels can indicate the degree of mitochondrial damage.24 25 We performed immunofluorescence staining of brain tissue and found more Parkin and Pink1 expression in the SAH group than the Sham group but no significant change in the Exo and Exo/Scr groups. When Exo/miR was given, these expression levels increased significantly (figure 2A-D) (p<0.05).

Exo/miR alleviates neuroinflammation by promoting mitophagy after SAH. A–D Immunofluorescence staining and quantitative analysis of Pink1 and Parkin in cortex 24 hours after SAH. Scale bar: 50 μm. E–I, Western blot and quantitative analysis of P62, Parkin, Pink1 and LC3 A/B in the mice (Sham, SAH, Exo, Exo/Scr and Exo/miR) groups. J,K Mitochondrial membrane potential and mitochondrial ROS production were analysed by TMRE and MitoSOX in the mice (Sham, SAH, Exo, Exo/Scr and Exo/miR) groups. L,M Representative TEM images of autophagosome and analysis after SAH mice model (The black triangle refers to the autophagosome and double black triangle refers to the autophagolysosome). Scale bar: 5 μm (upper panels), 1 μm (lower panels). **p<0.01, ***p<0.001, ****p<0.0001.
To verify this phenomenon, we measured the changes in mitophagy-related proteins. We found that Parkin, Pink1 and LC3 A/B protein expression moderately increased after SAH, and expression levels increased more significantly after Exo/miR treatment. By contrast, the P62 protein showed opposite results (figure 2E-I) (p<0.05). We also used primary microglia to measure the mitochondrial membrane potential and reactive oxygen species (ROS) levels by FCM. The SAH group had lower potential but higher ROS levels than the Sham group due to the depolarisation of mitochondrial membrane potential. After Exo/miR treatment, the membrane potential moderately recovered but remained lower than the Sham group. In contrast, MitoSOX showed the opposite result (figure 2J,K). These findings suggest that Exo/miR promotes mitophagy to clear damaged mitochondria and improve mitochondrial function, inhibiting the production of ROS and reducing neuroinflammation.
To understand the effect of Exo/miR on mitophagy, we conducted TEM observations in mouse brain microglia. The SAH group had more autophagosomes than the Sham group (figure 2L,M), and the number of autophagosomes increased significantly after Exo/miR treatment; some mitophagosomes were seen (double black triangle). These findings suggest that Exo/miR reduces neuroinflammation through mitophagy.
Exo/miR reduces neuroinflammation through SIRT2 inhibition of mitophagy
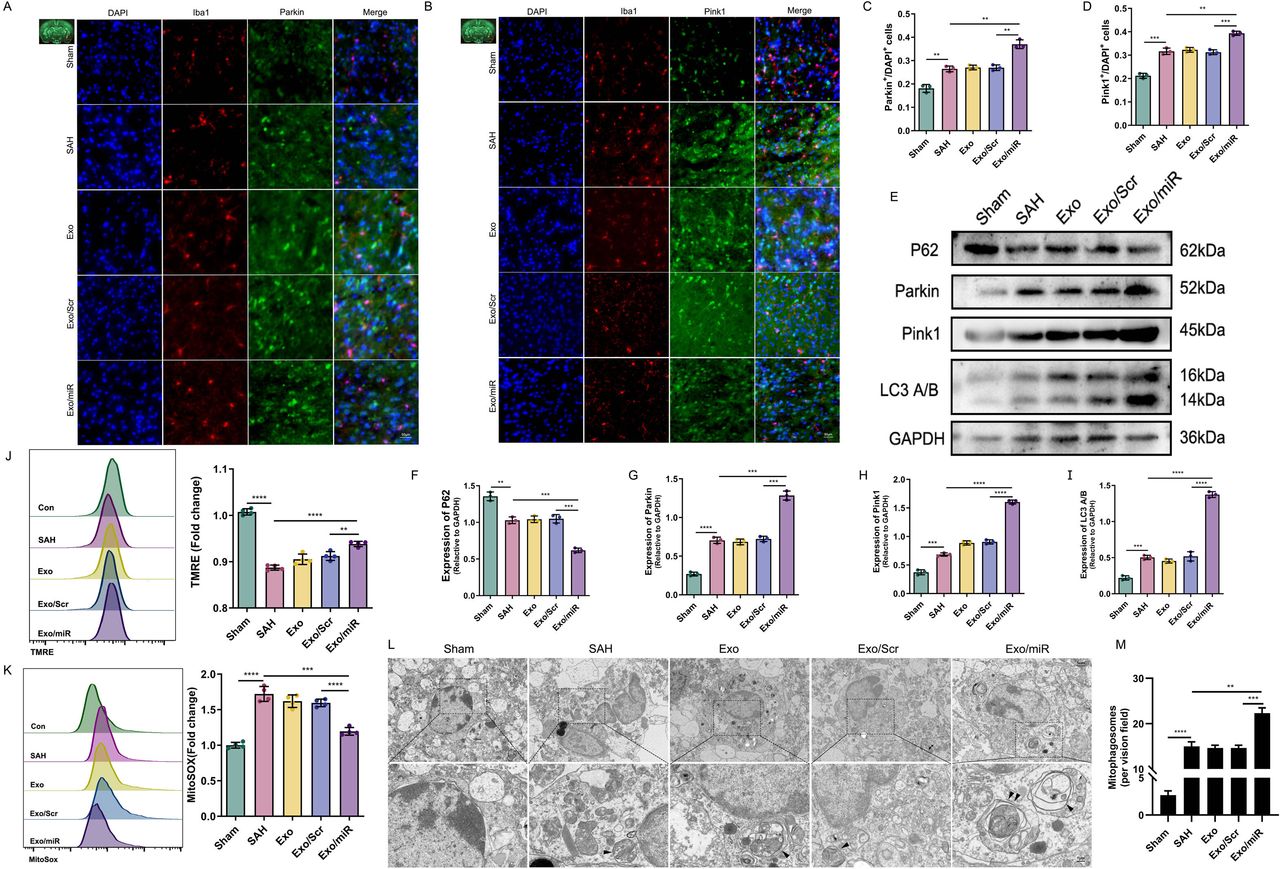
We conducted a bioinformatics analysis to identify potential targets for the effect of Exo/miR on mitophagy. We used miRanda (http://www.microrna.org/microrna/home.do), RNAhybrid (https://bio.tools/RNAhybrid/) and TargetScan (http://targetscan.org/) to predict the target genes of miR-486-3p. First, the 3′ UTR sequences of all protein-coding genes were obtained from the Ensembl database, and the sequence of miR-486-3p was obtained from miRbase. Then, three tools were run, and 4641, 4245 and 5265 target genes were obtained. Taking the intersection of three databases, we found that 1086 genes were identified using these tools. To identify the genes regulated by miR-486-3p in SAH, we obtained the gene expression profile of SAH from the GSE36791 database and analysed the expression of these genes.26 Using the limma package in R (V.4.2.0), we found that among these 1086 genes, only 12 were differentially expressed (|log2(fold-change) | >1, FDR <0.05). SIRT2 showed the largest fold-change among the upregulated genes. This finding was consistent with the expression pattern of miR-486-3p (figure 3A,B). To verify this result, we used TargetScan (8.0) to predict whether there is a binding site between miR-486-3p and SIRT2; surprisingly, there was indeed a binding site, which was verified by the luciferase experiment (figure 3C,D).

Exo/miR reduces neuroinflammation through SIRT2. A–D miR-486-3p bind with sirt2 was analysed and verified by bioinformatics and luciferase experiment. E,F Western blot and quantitative analysis of SIRT2 at 6 hours, 12 hours, 24 hours and 72 hours after SAH in mice. G–J Immunofluorescence staining and quantitative analysis of SIRT2 in cortex (G,H) and primary microglia 24 hours after SAH (I,J). Scale bar: 50 μm. K,L Western blot and quantitative analysis of SIRT2 in the mice (Sham, SAH, Exo, Exo/Scr and Exo/miR) groups. **p<0.01, ***p<0.001, ****p<0.0001.
We measured the expression of SIRT2 in SAH mice. SIRT2 expression differed at four time points; the expression level was the highest at 24 hours (figure 3E,F). Simultaneous immunofluorescence staining (24 hours) showed similar results (figure 3G-J). To explore the effects of Exo/miR and SIRT2 on mitophagy, we measured SIRT2 expression and found that it increased after SAH; however, SIRT2 levels decreased after treatment with Exo/miR (figure 3K,L). These findings suggest that SIRT2 may inhibit mitophagy.
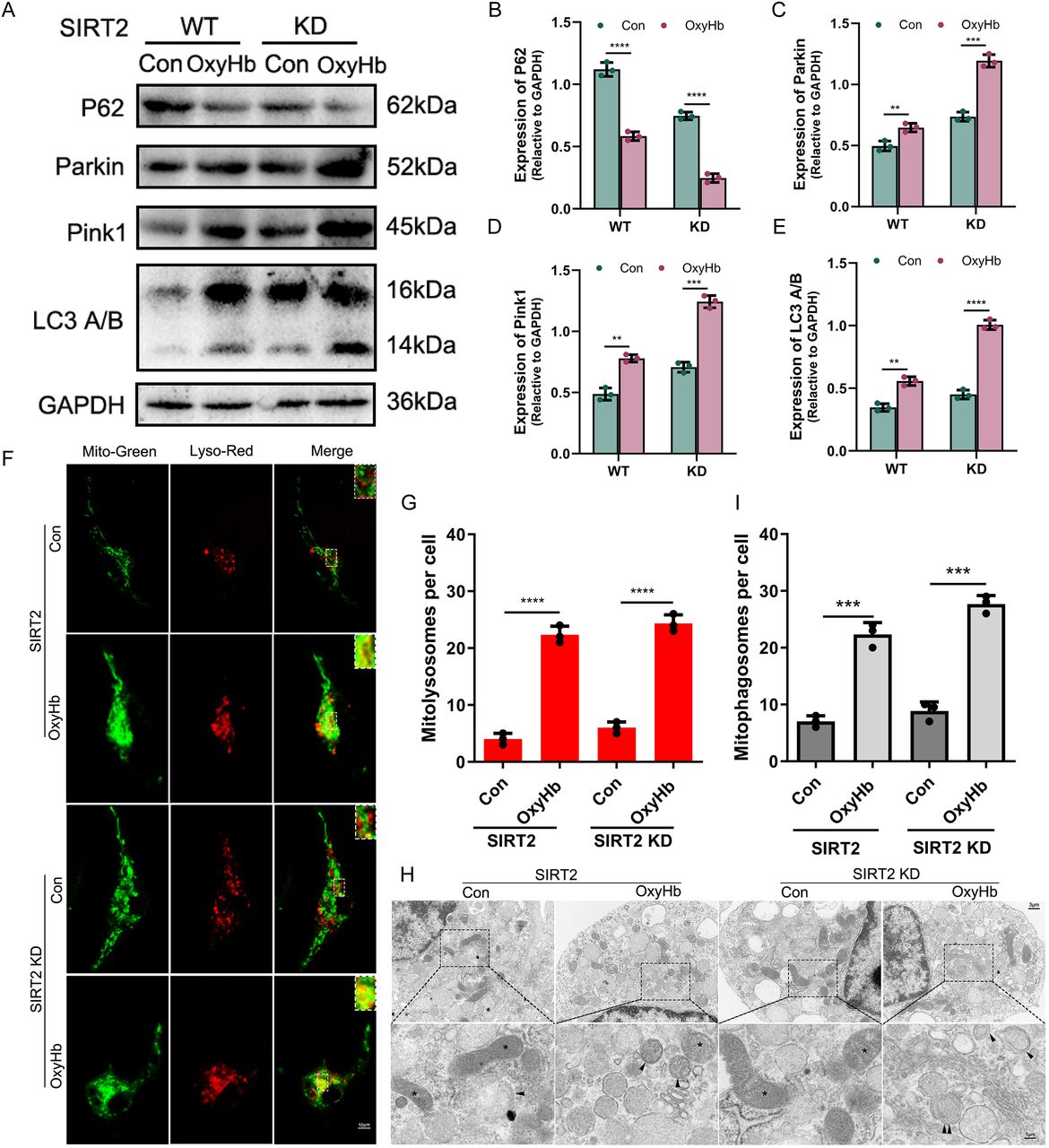
Then, we constructed a SIRT2 knockdown gene and a SAH cell model to test this hypothesis. First, we constructed SIRT2 knockdown microglia (SIRT2 KD) (online supplemental figure S4) and SAH models using OxyHb (25 µmol/L) and measured the level of mitophagy-related proteins. In the wild-type (WT) group, the western blot revealed that the expression of P62 decreased in the OxyHb group compared with the control group, while the opposite results were shown for Parkin, Pink1 and LC3 A/B. Interestingly, consistent results were also seen in the KD group; their expression levels were more evident than in the WT group (figure 4A-E) (p<0.05). These findings suggest that SIRT2 inhibits mitophagy activity in microglia. To test this hypothesis, we performed Mito-Tracker and Lyso-Tracker staining in the WT and KD groups, respectively. In both groups, there were significantly more mitolysosomes in the OxyHb group than in the Con group, and there were considerably more mitolysosomes in the KD group than in the WT group (figure 4F,G) (p<0.05). TEM was used to observe the effect of SIRT2 on mitophagy. The results were consistent with immunofluorescence staining; there were more mitophagosomes in the OxyHb group than in the Con group and significantly more mitophagosomes in the KD group than in the WT group (figure 4H,I) (p<0.05). These findings suggest that SIRT2 inhibits mitophagy activity in microglia.

SIRT2 inhibits mitophagy activity in microglia. A–E Western blot and quantitative analysis of P62, Parkin, Pink1 and LC3 A/B in SIRT2 WT and KD microglia. F,G The colocalisation of Mito-Green and Lyso-Red with laser confocal and quantitative analysis in different microglia. Scale bar: 10 μm. H,I Representative TEM images of autophagosome and quantitative analysis after SAH cell model (The black triangle refers to the autophagosome and double black triangle refers to the autophagolysosome and the * refers to mitochondria). Scale bar: 3 μm (upper panels), 1 μm (lower panels). **p<0.01, ***p<0.001, ****p<0.0001.
SIRT2 inhibits mitophagy through the deacetylation of PGC-1α after entering the nucleus
Exo/miR promotes mitophagy but inhibits mitochondrial autophagy after binding to SIRT2. Therefore, we speculated that there should be potential targets downstream of SIRT2 to regulate mitophagy. Because SIRT2 is found in the cytoplasm and mitochondria, we performed immunofluorescence staining on SIRT2 to determine any changes in its subcellular structure.27 28 Laser confocal imaging revealed that SIRT2 is primarily found in the cytoplasm; however, some SIRT2 entered the nucleus after stimulation with OxyHb (figure 5C). To clarify this phenomenon, we performed nucleocytoplasmic separation of cells. The western blot analysis showed that SIRT2 expression was higher in the nucleus than in the cytoplasm, and the proportion of SIRT2 was significantly higher after stimulation with OxyHb (figure 5A,B) (p<0.05). These findings suggest that SIRT2 may play a central role via nuclear translocation after SAH.

SIRT2 enter the nucleus and deacetylation of PGC-1α. A,B Western blot and quantitative analysis of SIRT2 in microglia (Con and OxyHb) group with Cyto and nucleus. C The colocalisation of SIRT2 and nucleus with laser confocal and analysis in Con group and OxyHb group of microglia. Scale bar: 10 μm. D Molecular docking shown an obvious interaction force between the protein PGC-1α and SIRT2. E,F Western blot and quantitative analysis PGC-1α in SIRT2 WT and KD microglia. G,H Coimmunoprecipitation and quantitative analysis of acetylated PGC-1α in SIRT2 WT and KD microglia. *p<0.05, ****p<0.0001, n.s., not significant.
However, it remained unclear whether SIRT2 affected mitophagy after entering the nucleus. Studies have found29 that SIRT2 regulated mitochondrial biogenesis, upregulated antioxidant enzyme expression and reduced ROS levels by regulating PGC-1α. Then, we explored the relationship between SIRT2 and PGC-1α. First, we downloaded the protein (SIRT2) structure file (PDB: AF-Q8IXJ6-F1) and the PGC-1α structure file (PDB: AF-Q9UBK2) from PDB (https://www.uniprot.org/blast/). Then, we used Python (V.3.7.7)-Pymol (V.2.4.0) for docking graphics processing and intermolecular spatial distance (a distance less than 5 Å indicates an evident interaction force). Molecular docking results showed hydrophobic forces between PGC-1α- GLU655 and SIR2- ALA198(3.1 Å) and PGC-1α- GLN388 and SIRT2- ALA241 (5.0 Å). These findings suggest an interaction force between the protein PGC-1α interacting with SIRT2 and possible interaction in space (figure 5D).
We explored the effect of SIRT2 on PGC-1α. As shown in figure 5E,F, the expression level of PGC-1α showed almost no change while SIRT2 KD, suggesting that SIRT2 does not directly affect PGC-1α. We next determined whether SIRT2 affected PGC-1α; considering that SIRT2 is a deacetylase, we determined whether there is acetylation modification on PGC-1α. Therefore, we performed acetylation co-immunoprecipitation experiments on PGC-1α. As we hypothesised, acetylation modification exists between SIRT2 and PGC-1α, and SIRT2 KD significantly increased the acetylation level of PGC-1α (figure 5G,H) (p<0.05). These findings suggest that SIRT2 acts by reducing the acetylation level of PGC-1α.
Next, we determined how PGC-1α influences mitophagy. First, we constructed PGC-1α knockdown microglia (PGC-1α KD) (online supplemental figure S5) and SAH models using OxyHb (25 µmol/L). Then, we divided microglia into Con, Hb, Hb+SIRT2 KD, Hb+PGC-1α KD and Hb+SIRT2 KD+PGC-1α KD groups to explore the potential mechanism. The western blot analysis revealed no significant difference between the P62 protein expression level in the Hb group and Hb+PGC-1α KD group, and it was slightly lower than in the Con group. However, the expression level in the Hb+SIRT2 KD group was significantly lower. When SIRT2 and PGC-1α were KD, P62 levels were elevated but still lower than the Hb and Hb+PGC-1α KD groups. By contrast, Parkin, Pink1 and LC3 showed the opposite results. The protein levels between Hb and Hb+PGC-1α KD were not significantly different and were moderately increased compared with the Con group; however, the expression level of the Hb+SIRT2 KD group increased significantly. Although their expression levels decreased when SIRT2 and PGC-1α were KD, they were slightly higher than in the Hb and Hb+PGC-1α KD groups (figure 6A-E). These findings suggest that mitophagy decreases after PGC-1α KD, and PGC-1α promotes mitochondrial autophagy.

PGC-1α promotes mitophagy activity in microglia. A–E Western blot and quantitative analysis of P62, Parkin, Pink1 and LC3 A/B in the microglia (Con, Hb, Hb + SIRT2 KD, Hb + PGC-1α KD and Hb +SIRT2 KD + PGC-1α KD) groups. F,G The colocalisation and analysis of Autophagosome and Mito-Red with laser confocal in different groups. Scale bar: 10 μm. H,I The colocalisation of Mito-Green and Lyso-Red and analysis with laser confocal in different groups. Scale bar: 10 μm. J,K Representative TEM images of autophagosome after SAH cell model (The black triangle refers to the mitolysosome and double black triangle refers to the autophagolysosome and the * refers to mitochondria). Scale bar: 3 μm (upper panels), 1 μm (lower panels). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s., not significant.
To confirm this result, we used Autophagosome+Mito-Tracker Red for immunofluorescence staining to observe mitophagosomes and Mito-Tracker Green+Lyso-Tracker Red to observe mitolysosomes. Compared with the Con group, the levels of mitophagosomes and mitolysosomes in other groups were more significant, with the highest level in the Hb+SIRT2 KD group. Compared with the Hb+SIRT2 KD group, the number of mitophagosomes and mitolysosomes was significantly lower after PGC-1α KD (figure 6F-I) (p<0.05). This finding suggests that PGC-1α promotes mitophagy. TEM also revealed these differences (figure 6J,K), suggesting that PGC-1α promotes mitophagy.
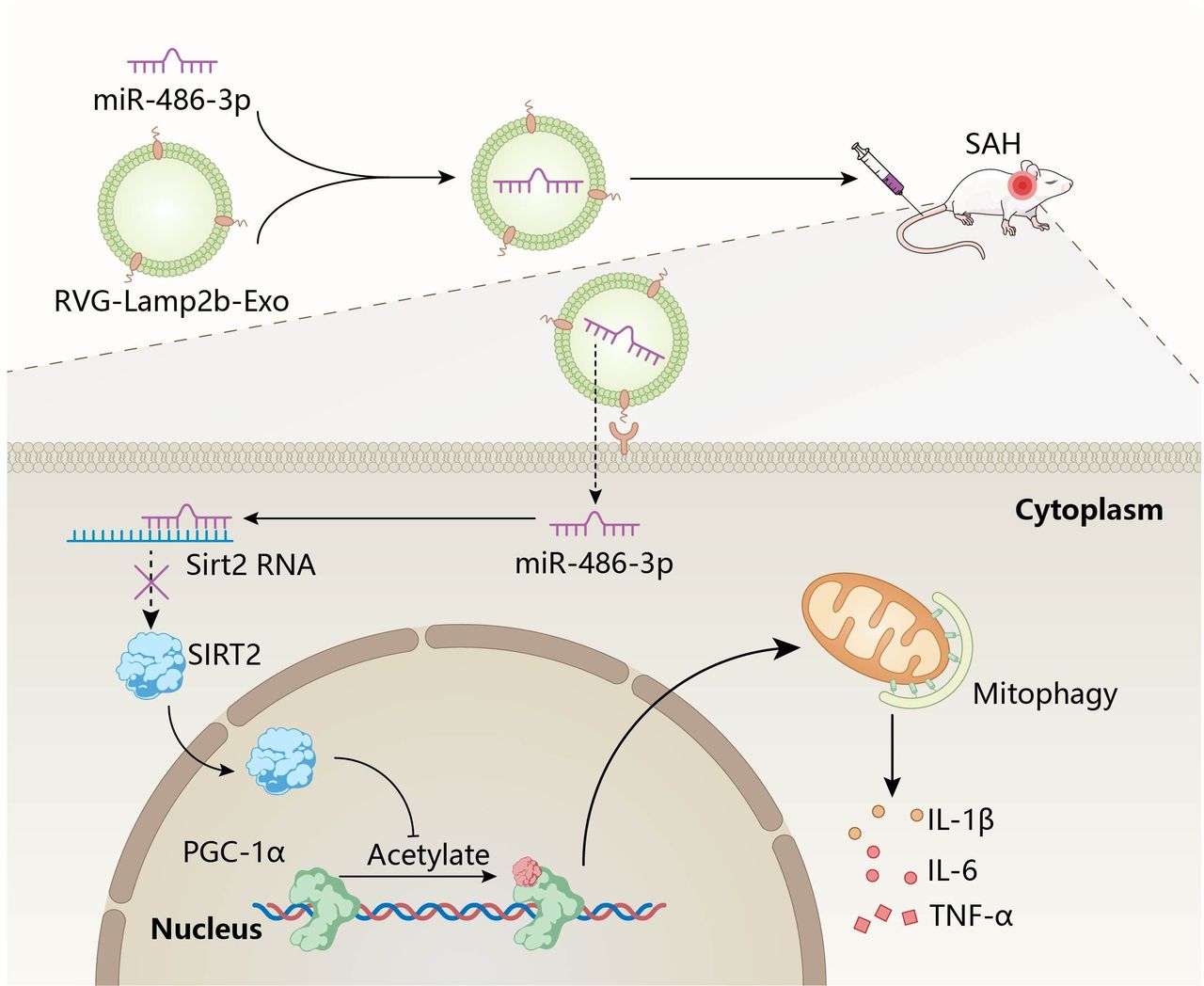
Then, we determined the role of Exo/miR in SAH. We engineered Exo into RVG-Lamp2b to target them to the brain; then, miR-486-3p was loaded into the engineered exosomes through electroporation to construct Exo/miR. Finally, the Exo/miR was injected into the SAH mice through the tail vein to target the periphery of brain tissue. Then, miR-486-3p is released and combined with SIRT2 to cause phenotypic conversion of microglia. SIRT2 enters the nucleus and regulates the level of mitophagy by inhibiting the acetylation level of PGC-1α, alleviating neuroinflammation (figure 7).

Schematic illustration of the effect of Exo/miR on alleviate neuroinflammation in SAH mice. Intravenous administration of Exo/miR following SAH treatment alleviate neuroinflammation through microglia occurs a phenotypic conversion. miR-486-3p binds to sirt2 and downregulates downstream gene PGC-1α and inhibits mitophagy to alleviate neuroinflammation.
Discussion
In the present study, we targeted the delivery of miR-486-3p to the brain via engineered RVG-Exo and demonstrated that Exo/miR improved neuroinflammation in SAH mice. These Exo alleviated neuroinflammation and reduced brain tissue oedema and BBB damage after SAH by regulating microglia phenotype conversion and mitophagy. Functional studies showed that miR-486-3p binds to SIRT2, which regulates the acetylation of PGC-1α when entering the nucleus, alleviating neuroinflammation after SAH by regulating mitophagy.
The development of medications to treat SAH is limited by complex challenges posed by the neurovascular unit. The BBB strictly controls the entry of small molecules and cells into the brain, restricting pathogenic factors and immune cells from blood vessels from reaching the brain and limiting medication delivery.30 31 Conventional approaches using free forms of drug molecules show poor BBB penetration because efflux pumps on endothelial cells tightly regulate the movement of molecules.32 33 In recent years, studies have shown that specific ligands such as RVG can be expressed on the membrane surface of exosomes through chemical/gene modification, enabling targeted delivery of exosome. Haroon K et al34 modified exosomes with neuron targeting peptide RVG29 via bio-orthogonal click chemistry technique and loaded it with NR2B9c, delivering RVG-ExoNR2B9c to brain and alleviating damage after traumatic brain injury. In the ischaemic stroke model, Haroon K et al used the same approach to couple RVG29 with the surface of exosomes and load them with NR2B9c, ultimately generating stroke-specific therapeutic COCKTAIL.35 Yu et al36 loaded circDYM into RVG-EVs to achieve targeted delivery to the brain of CUS mice model and alleviate CUS-induced depressive-like behaviour. Therefore, we can achieve targeted delivery miR-486-3p to the brain by modifying exosomes.
Several lines of evidence suggest that miR-486-3p has different biological functions in the central nervous system. Exosomal miR-486-3p induced by bone marrow mesenchymal stem cells prevents diabetic retinopathy through TLR4/NF-κB axis inhibition37; miR-486-3p enhanced the sensitivity of temozolomide in glioblastoma by targeting O6-methylguanine-DNA methyltransferase (MGMT).38 Deep sequencing of extracellular vesicle small RNA showed that miR-486-3p was a circulating element distinguishing patient with glioblastoma from those with low-grade astrocytoma.39 In addition to its function as a miRNA sponge, it can bind to RNA-binding proteins and affect their distribution and function. Studies demonstrated that miR-486-3p binds with RNA-binding proteins, affecting their distribution and function.40 41
In the present study, using bioinformatics prediction, analysis and dual-luciferase experiments, we found that miR-486-3p regulates microglia phenotypes after binding to SIRT2 and reduces neuroinflammation by regulating mitophagy after SAH. SIRT2 is an NAD+-dependent protein deacetylase critical in neurological diseases and participates in pathological changes, including autophagy and neuroinflammation.42 43 Studies indicated that SIRT2 enhances NAD/NADH-mediated ATP increase in microglia and prevents inflammation induced by excessive microglial activation through deacetylation of NF-κB.44 By contrast, SIRT2 inhibition enhances the acetylation of NF-κB, reduces its phosphorylation at Ser331 and Ser335, and upregulates AQP4 and MMP-9, which causes SIRT2 to undergo nuclear translocation, promoting microglia activation and pro-inflammatory cytokine expression, exacerbating neuroinflammation.45
Krishnan et al29 reported that SIRT2 regulates mitochondrial biogenesis and upregulates the expression of antioxidant enzymes, reducing ROS levels by regulating PGC-1α. PGC-1α regulates SIRT2 and is localised in the cytoplasm and nucleus. Its cellular localisation depends on its acetylation status, and it prefers the nucleus during oxidative stress.46 SIRT2 undergoes nuclear translocation when it is inhibited by changing the acetylation levels of mitochondrial proteins and inhibiting mitophagy through PGC-1α.47 Our study found that SIRT2 inhibition increased the acetylation level of PGC-1α, and SIRT2 reduced the acetylation level of PGC-1α. PGC-1α can promote mitophagy in microglia and inhibit neuroinflammation.48 In fact, the connection between microglia polarisation and mitophagy has been reported. Zhang et al demonstrated49 that an inhibited HMGB1/RAGE axis led to increased mitophagy, which could reduce the activity of microglia-mediated neuroinflammation. Yang et al reported50 that melatonin, through AMPKα2-mediated mitophagy enhancement, facilitates microglia polarisation toward the M2 phenotype and alleviates LPS-induced neuroinflammation. These findings suggest mitophagy may affect microglia polarisation and phenotype conversion, alleviating neuroinflammatory damage. Therefore, we believe that SIRT2 reduces neuroinflammation by inhibiting mitophagy and microglial polarisation and regulating the acetylation level of PGC-1α. Consistent with these findings, we demonstrated that miR-486-3p with SIRT2 causes phenotypic switching of microglia and reduces neuroinflammation by regulating mitophagy after SAH; mitophagy was regulated by reducing the acetylation level of PGC-1α after SIRT2 enters the nucleus.
Although this study revealed the targeted delivery of RVG-Exo and the potential mechanism of miR-486-3p in neuroinflammation after SAH, there were some limitations. First, despite substantial progress in exosome-based drug delivery systems, there is no ideal purification technology to isolate and purify exosomes with high purity, limiting the clinical application of exosome-based drug delivery.51 Second, although SIRT2 is a potential target of miR-486-3p in the SAH mice brain model, it was not the unique target, which may be regulated by other mechanisms. Finally, although Exo/miR could cross the BBB, the BBB damage will affect its effect to a certain extent after SAH. Therefore, we should fully consider this in future experimental designs or use other administration methods to maximise its impact.
Conclusions
RVG-Exo improve the efficiency of miR-486-3p delivery, which can reduce neuroinflammation by inhibiting SIRT2 expression and increasing mitophagy after SAH. These findings suggest that Exo-based delivery of miR-486-3p is a promising therapeutic strategy for SAH.
Data availability statement
Data are available on reasonable request from CHH.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the Ethics Committee of the Nanjing Drum Tower Hospital (2021–313-07). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank all the other members in their laboratory for their insight and technical support. We also thank to the patients who participate this research.
Footnotes
BS, SG, XC and YL contributed equally.
LW, C-HH and WL contributed equally.
Contributors All authors contributed to the study conception and design. LYW, WL and CHH conceived and designed the research; SG, JQS and YZ performed experiments; LYW analysed the data; BS, XXC, YL and JD drafted the manuscript; all the authors reviewed and approved the final version of the manuscript. CHH is responsible for the overall content.
Funding This work was supported by grants from the National Natural Science Foundation of China (No. 82271363(LY Wu), 82301485(XX Chen), 82201456(Y Zhou), 82130037(CH. Hang)), Natural Science Foundation of Jiangsu Province (No. 2022JJ30060(Y Zhou)), Anhui Provincial Department of Education Excellent Scientific Research and Innovation Team Project in Colleges and Universities (2022AH010073(NS Lai)) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX24_0010(B Sheng)).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
References


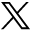


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}