

Abstract
Objective This study aims to investigate the prevalence of familial cerebral cavernous malformations (FCCMs) in first-degree relatives (FDRs) using familial screening, to describe the distribution of initial symptoms, lesion count on cranial MRI and pathogenic gene in patients.
Methods Patients with multiple CCMs who enrolled from the Treatments and Outcomes of Untreated Cerebral Cavernous Malformations in China database were considered as probands and FDRs were recruited. Cranial MRI was performed to screen the CCMs lesions, and whole-exome sequencing was performed to identify CCM mutations. MRI and genetic screening were combined to diagnose FCCM in FDRs, and the results were presented as prevalence and 95% CIs. The Kaplan-Meier (KM) method was used to calculate the cumulative incidence of FCCM.
Results 33 (76.74%) of the 43 families (110 FDRs) were identified as FCCM (85 FDRs). Receiver operating characteristic analysis revealed three lesions on T2-weighted imaging (T2WI) were the strong indicator for distinguishing probands with FCCM (sensitivity, 87.10%; specificity, 87.50%). Of the 85 FDRs, 31 were diagnosed with FCCM, resulting in a prevalence of 36.5% (26.2%–46.7%). In families with FCCMs, the mutation rates for CCM1, CCM2 and CCM3 were 45.45%, 21.21% and 9.09%, respectively. Furthermore, 53.13% of patients were asymptomatic, 17.19% were intracranial haemorrhage and 9.38% were epilepsy. The mean age of symptom onset analysed by KM was 46.67 (40.56–52.78) years.
Conclusion Based on MRI and genetic analysis, the prevalence of CCMs in the FDRs of families with FCCMs in China was 36.5%. Genetic counselling and MRI screening are recommended for FDRs in patients with more than three CCM lesions on T2WI.
WHAT IS ALREADY KNOWN ON THIS TOPIC
Familial cerebral cavernous malformations (FCCMs) exhibit an autosomal dominant inheritance pattern.
FCCM arises from loss of function mutations of three known genes: CCM1/KRIT1, CCM2/MGC4607 and CCM3/PDCD10.
WHAT THIS STUDY ADDS
The prevalence of CCMs in the first-degree relatives (FDRs) of families with FCCMs in China was 36.5%.
Most patients with FCCM in Chinese populations were known mutations.
About half of the patients with were asymptomatic and needed to be detected using screening.
More than three lesions on T2-weighted imaging (T2WI) are likely to FCCMs.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Genetic counselling and MRI screening are recommended for FDRs in patients with more than three CCM lesions on T2WI.
Introduction
Familial cerebral cavernous malformations (FCCMs) are the rare, genetic cerebrovascular disease, affecting approximately 1/3300–1/10 000 individuals based on the Orpha net (https://www.orpha.net/) and screening exome sequencing databases.1 2 In China, approximately 14 000–42 424 patients with FCCMs are estimated in a population of 1.4 billion. FCCMs have been reported to cause clinical symptoms, including epilepsy, intracranial haemorrhage (ICH), focal neurological deficits (FNDs) and headache, resulting in severe physical, psychological and financial difficulties in the family.3–6 Among those affected, 20%–50% of patients with FCCMs remain asymptomatic, which emphasises their genetic risk.2 Therefore, the use of effective screening strategies is necessary, such as MRI and genetic screening.
FCCM exhibits an autosomal dominant inheritance pattern,7 8 indicating that 50% of the first-degree relatives (FDRs) will inherit this germline variant theoretically. The prevalence of FCCM in FDRs is difficult to be estimated due to incomplete penetrance and inconsistent presentation of the disease.2 Genetic mutations and their MRI and clinical manifestations are complex in the real world. However, there are limited data on the CCM prevalence in FDRs among families with FCCMs. Furthermore, FCCM arises from loss of function mutations of three known genes: CCM1/KRIT1, CCM2/MGC4607 and CCM3/PDCD10.7 8 The distributions of these pathogenic genes vary widely in the literature, denoting differences due to ethnic factors. Particularly, large-scale genetic studies of the Chinese population are crucial to describe this specific distribution of pathogenic genes. The findings of such studies can provide evidence-based support for clinical genetic diagnosis, genetic counselling and genetic intervention in Chinese patients with FCCMs.
Given these circumstances, this study aimed to analyse the prevalence of CCMs in FDRs and describe the clinical and genetic characteristics of patients. MRI was conducted to screen patients with multiple CCMs and their FDRs, and whole-exome sequencing (WES) was performed for all members with CCMs.
Methods
Study design and participants
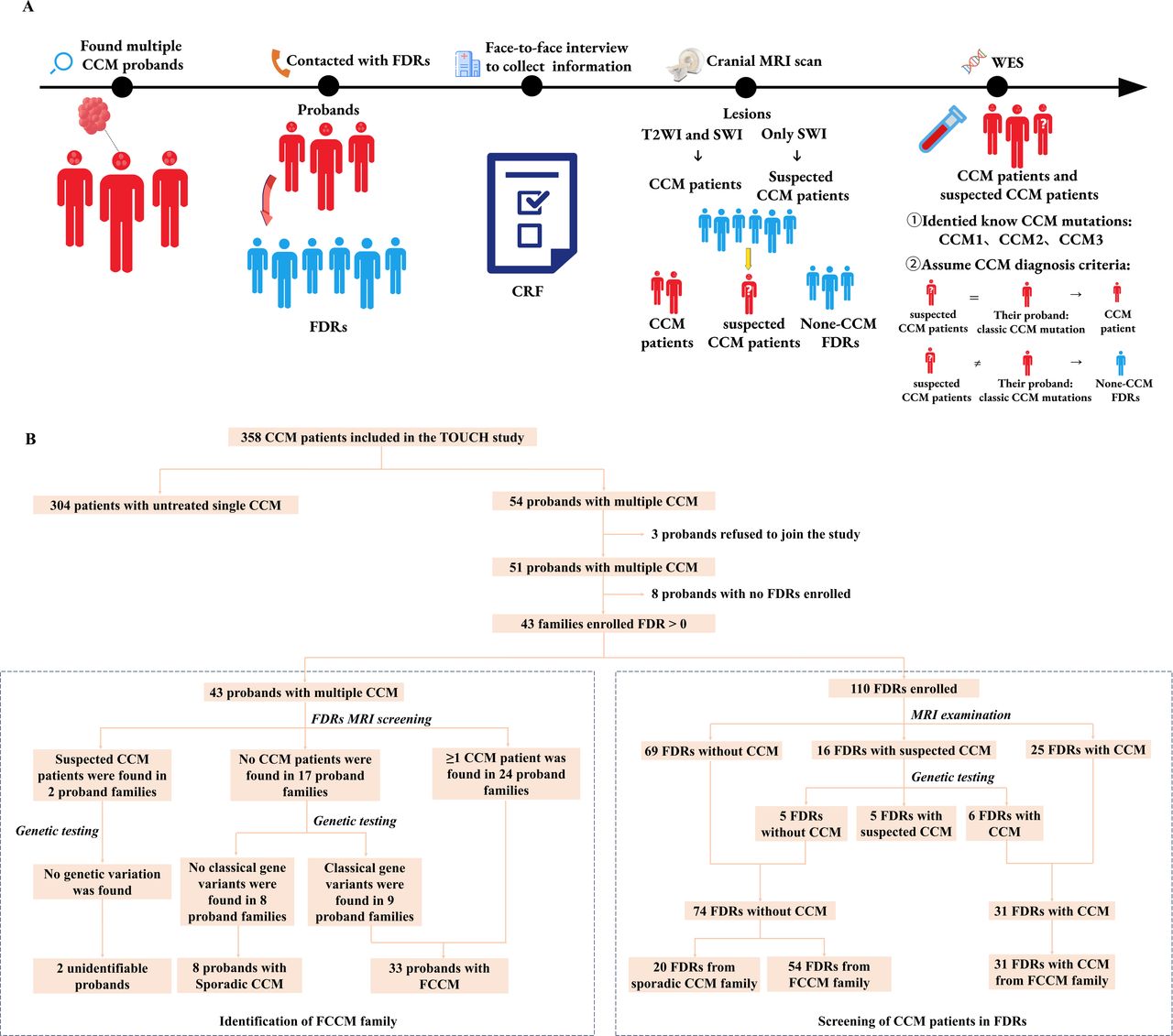
The Treatments and Outcomes of Untreated Cerebral Cavernous Malformations in China (TOUCH) study (Clinical Trials Registry of National Institutes of Health, registration ID: NCT03467295) was a nationwide, multicentre, prospective cohort study that enrolled untreated single or multiple CCM lesions or partially treated patients with multiple CCM lesions (having untreated CCM lesions). Between November 2020 and May 2023, we enrolled patients with multiple CCM lesions in our centre from the TOUCH database and their respective FDRs. The study design and process are shown in figure 1A. Multiple CCMs were defined as having at least one CCM lesion according to Zabramski’s diagnostic criteria9 and ≥2 lesions on cranial susceptibility weighted imaging (SWI). FDRs who were screened included parents, siblings and offsprings. Basic participant information was collected via face-to-face interviews, and cranial MRI was performed for all participants. Based on screening results, the FDRs were then categorised into the CCM, assumed CCM and non-CCM groups. Lastly, blood samples were collected from all participants, and WES was performed for patients in the CCM and assumed CCM groups to identify known CCM mutations (CCM1, CCM2 and CCM3) and confirm screening findings.

Study design and screening process for FDRs of patients with FCCMs. (A) Study design and workflow. (B) FCCM families and FDRs were identified and screened. The dotted box on the left demonstrates the 43 multiple CCM probands for identifying FCCM families by the integration of cranial MRI and WES. The dotted box on the right demonstrates the process of diagnosing CCM by cranial MRI combined with WES in 110 FDRs of multiple CCM probands. FCCMs, familial cerebral cavernous malformations; FDRs, first-degree relatives; CRF, case report form; WES, whole-exome sequencing.
Our manuscript was structured in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology reporting guidelines for cohort studies.10
Screening of participants
All participants underwent cranial MRI, blood sample collection and interviews on enrolment. Cranial MRI scans, including T1-weighted imaging (T1WI), T2WI and SWI, were performed on patients with multiple CCMs and FDRs. Lesions with a maximum diameter of at least 4 mm on T2WI were counted to determine the total number of lesions. Using the collected blood samples, WES was performed for all patients with CCMs to identify the pathogenic gene and mutation type. Pathogenic mutation was identified according to the American College of Medical Genetics and Genomics (ACMG) guidelines.11 Novel mutations were defined as those absent from the Human Gene Mutation Database (HGMD, https://www.hgmd.cf.ac=/) and ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) at the time of analysis. A trained epidemiologist or neurosurgeon conducted face-to-face interviews with each participant to collect basic information, including general demographics, initial symptoms, medical history and family history. Initial symptoms were defined as those experienced at the time of CCM first detection on cranial MRI, including ICH, epilepsy without ICH, FNDs without ICH and others (eg, headaches, dizziness). All data were recorded in the EDC database (Real Data EDC system) using a ‘one-person input, one-person verification’ approach to ensure data accuracy for subsequent analysis.
WES and analyses
Peripheral blood samples were used to generate the DNA library, as described in a previous study.12 The whole exome was captured using the SureSelect Human All Exon Kit V6 (Agilent Technologies, USA) or IDTxGenExome2 kit (Integrated DNA Technologies, USA). The target region was sequenced using the NovaSeq 6000 platform (Illumina, USA) with high throughput, achieving an average coverage depth of more than 80×and at least 94% of target exons covered above 20×depth. The GRCh37/hg19 were used as human reference genomes in this study. Paired-end sequence reads, realignment, duplicate removal and call variants were performed using Burrows-Wheeler Aligner, SAMtools, Picard and Haplotype Caller of the Genome Analysis Toolkit (GATK) software according to the standard operating procedure (SOP). Pathogenic genes and mutations were identified according to known CCM phenotypes and established guidelines from the ACMG.
FCCM diagnosis criteria
CCM diagnosis in FDRs was based on the following criteria: (1) the presence of low-sign lesions on SWI that fulfil Zabramski’s diagnostic criteria on T2WI and T1WI9 or (2) the presence of low-signal lesions on SWI alone. For the latter criterion, patients were defined as those with assumed CCM, and further testing was performed with WES. If CCM gene mutations were consistent with those in their probands, these patients would now be defined as definite CCM cases. For patients with CCMs, FCCM diagnosis was based on the following criteria: (1) identification of a known pathogenic CCM mutation or (2) the presence of at least two patients with CCMs within the family.
Quality assurance
Clinical information was collected by trained neurosurgeons and epidemiologists using a customized case report form (CRF). Cranial MRI scans were performed by two trained radiographers using the same MRI machine and parameters. Acquisition parameters for T2WI (repetition time (TR), 4000 ms; echo time (TE), 103 ms; field of view (FOV), 200×230 mm; matrix, 340×340; slices, 22; slice thickness, 5 mm) and SWI (TR, 31 ms; TE, 7.2 ms; FOV, 200×230 mm; matrix, 384×332; slices, 130; slice thickness, 2 mm) were standardised. CCM diagnosis and lesion count on MRI were performed by a neurosurgeon and a neuroradiologist after reaching a consensus. Any inconsistencies were resolved by a senior neurosurgeon.
Statistical analysis
All statistical analyses were performed by using the SPSS Statistics software (V.5.0; SPSS) and R language software (V.4.1.0; Institute for Statistics and Mathematics, Vienna, Austria). Categorical data were presented as counts and proportions (n, %) and analysed using the χ2 test (continuity correction and Fisher’s exact test if necessary). Meanwhile, continuous variables were presented as means and standard deviation (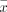 ±SD) or median and interquartile range (M(P25-P75)) and were analysed using either Student’s t-test (for normal distribution) or Mann-Whitney U test (for skewed distribution). The 95% CIs of prevalence were calculated by the normal approximation method. The receiver operating characteristic (ROC) was drawn and the area under the curve (AUC) value was calculated to evaluate the predictive ability of the number of lesions for the type of CCM, wherein the optimum cut-off value was determined using the Maximum Youden index method. The cumulative probability and mean age of symptoms onset in patients with CCMs were evaluated using the Kaplan-Meier (KM) method. Furthermore, the log-rank method was used to compare cumulative incidence between groups, and the Benjamini-Hochberg (BH) method was used for pairwise comparisons. All tests were two sided, and a p<0.05 was considered statistically significant.
±SD) or median and interquartile range (M(P25-P75)) and were analysed using either Student’s t-test (for normal distribution) or Mann-Whitney U test (for skewed distribution). The 95% CIs of prevalence were calculated by the normal approximation method. The receiver operating characteristic (ROC) was drawn and the area under the curve (AUC) value was calculated to evaluate the predictive ability of the number of lesions for the type of CCM, wherein the optimum cut-off value was determined using the Maximum Youden index method. The cumulative probability and mean age of symptoms onset in patients with CCMs were evaluated using the Kaplan-Meier (KM) method. Furthermore, the log-rank method was used to compare cumulative incidence between groups, and the Benjamini-Hochberg (BH) method was used for pairwise comparisons. All tests were two sided, and a p<0.05 was considered statistically significant.
Results
Identification of familial CCM and proband characteristics
Initially, 54 patients with multiple CCMs were enrolled from the TOUCH database. Three patients were excluded due to refusal of informed consent or blood sampling. The enrolment process is illustrated in figure 1B. Ultimately, 43 multiple CCM patients with at least one FDR inclusion (84.31%) were included in the final analysis. Based on cranial MRI and genetic screening, 33 families (76.74%) were classified as FCCM families, and 8 patients were identified to have sporadic CCM. Notably, two families had unclear CCM types due to the absence of known mutations and the presence of only one suspected CCM case among FDRs.
Clinical characteristics of the probands are shown in online supplemental table 1. The mean age at enrolment was 38.97±17.79 years in FCCM probands and 44.63±14.08 years in sporadic CCM probands. For SWI, the lesion number was ≥5 in 93.1% of FCCM probands, which was higher than that in sporadic CCM probands (12.5%; χ2 test, p<0.001). For T2WI, the FCCM probands exhibited a greater median number of lesions ≥4 mm compared with sporadic CCM patients (6 (4–10) vs 1.5 (1–2), p=0.001). Moreover, 45.2% of FCCM probands and 0% of sporadic CCM probands had more than 10 lesions ≥5 mm (sporadic CCM patients, 0%; χ2 test, p=0.034). Univariate analysis further revealed no statistically significant differences in the remaining variables between the two groups.
Supplementary data
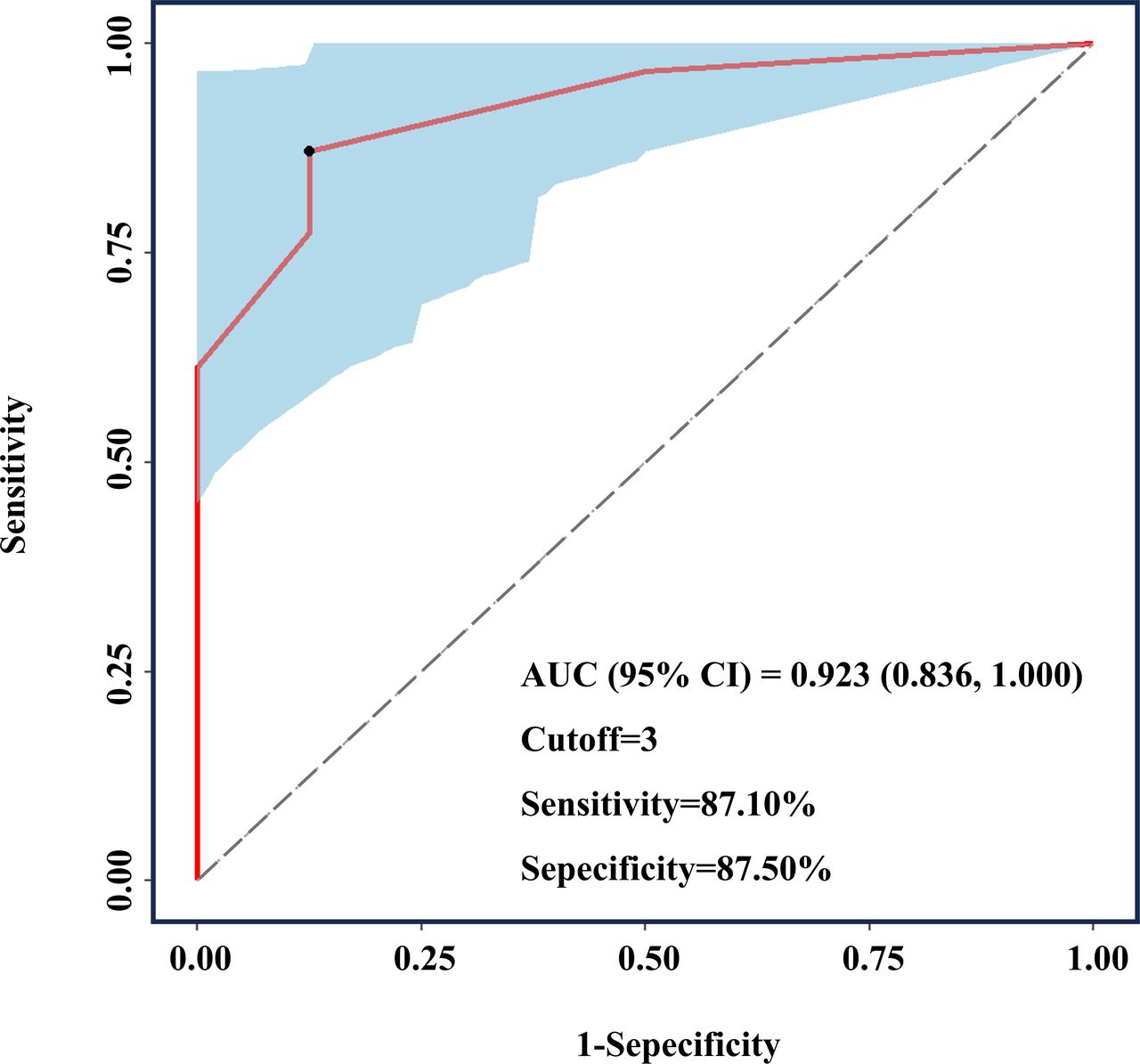
ROC curve analysis was performed to explore the ability of lesion count (≥4 mm on T2WI) to distinguish between patients with sporadic and familial CCMs. Figure 2 illustrates the ROC curves, with CCM type as the dependent variable and lesion number as the independent variable. Notably, the AUC value was 0.923 (0.836, 1.000). The optimal cut-off point of 3 was obtained at the maximum Youden index, with a sensitivity and specificity of 87.10% and 87.50%, respectively (figure 2).

ROC curve for distinguishing patients with FCCMs patients from those with multiple sporadic CCMs by number of the lesions T2WI. The AUC was 0.923 (0.836, 1.000), and the cut-off value based on the maximum Youden index was 3. The sensitivity was 87.10%, and the specificity was 87.50%. AUC, area under the curve; FCCMs, familial cerebral cavernous malformations; ROC, receiver operating characteristic; T2WI, T2-weighted imaging.
Prevalence and general demographics of FDRs in families with FCCMs
A number of FDRs enrolled in each family are shown in table 1 and online supplemental figure 1. A total of 110 FDRs from 43 probands with multiple CCMs were invited for screening. Of these, 85 FDRs from 33 (76.74%) FCCM families were included, and their basic information is shown in table 2. The CCM diagnostic process for FDRs is shown in figure 1B. Based on MRI, 25 FDRs were diagnosed with CCM, whereas 16 FDRs were categorised as assumed CCM. Among the latter, 6 FDRs were confirmed to have CCM due to positive WES findings that were consistent with their probands (online supplemental table 2). Finally, 31 FDRs were diagnosed with FCCM in this study, resulting in a prevalence of 36.5% (26.2%–46.7%). Including the five self-reported FDRs who underwent prior MRI but did not participate in the field investigation, the prevalence increased to 38.9% (28.8%–49.0%). To further explore the effect of the FDR enrolment rate on prevalence, we excluded families with FDR enrolment rates of less than 100% for sensitivity analysis. Among the 33 families with FCCMs, 11 families had an FDR enrolment rate of 100%, yielding a prevalence of 38.9% (23.0–54.8). The prevalence of FDRs in FCCM among different populations is shown in table 2.
Enrolment of FDRs of the proband
Characteristics of FDRs of probands with FCCM and prevalence of FDRs of FCCM
Pathogenic genes and clinical characteristics of patients with FCCMs
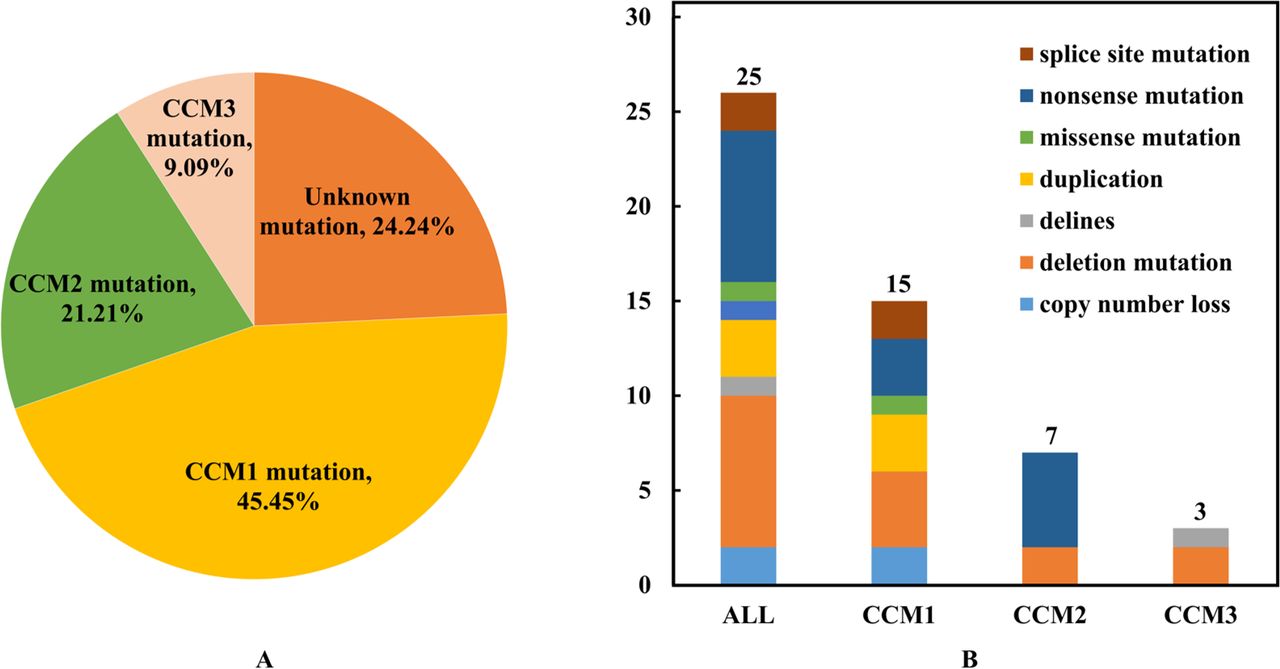
In the 33 FCCM families, the mutation rates of CCM1, CCM2 and CCM3 were 45.45% (15/33), 21.21% (7/33) and 9.09% (3/33), respectively (figure 3A). The remaining 24.24% (8/33) showed no identifiable pathogenic genes. Genetic analysis revealed eight mutation types and 22 mutation sites in CCM genes (figure 3B). Among them, CCM2 rs755800734 had the highest mutation rate, accounting for 16% (4/25) of all single nucleotide polymorphisms (SNPs) and 57.14% (4/7) of CCM2 SNPs (online supplemental table 3). Moreover, 10 novel mutations were identified in this cohort (online supplemental table 3), including our previously reported CCM1 mutation NG_012964.1 (NM_194456.1): c.1255–1G>T (splice-3).13

The distribution and types of CCM mutations in families with FCCMs. (A) Distribution of CCM mutations among 33 families with FCCMs. (B) Mutation types of the three classical CCM genes. FCCMs, familial cerebral cavernous malformations.
Clinical characteristics of patients with FCCMs (33 probands and 31 FDRs patients) and their different pathogenic genes are shown in online supplemental table 4. The proportion of CCM lesions on T2WI differed significantly among the four groups (p=0.041). Pairwise comparison further showed that patients with CCM3 mutations had a higher proportion of lesions on T2WI than those in the other three groups (all PBonferroni<0.05). Conversely, no statistically significant differences in the remaining variables were observed across the four groups.
Initial symptoms and cumulative incidence in patients with FCCMs
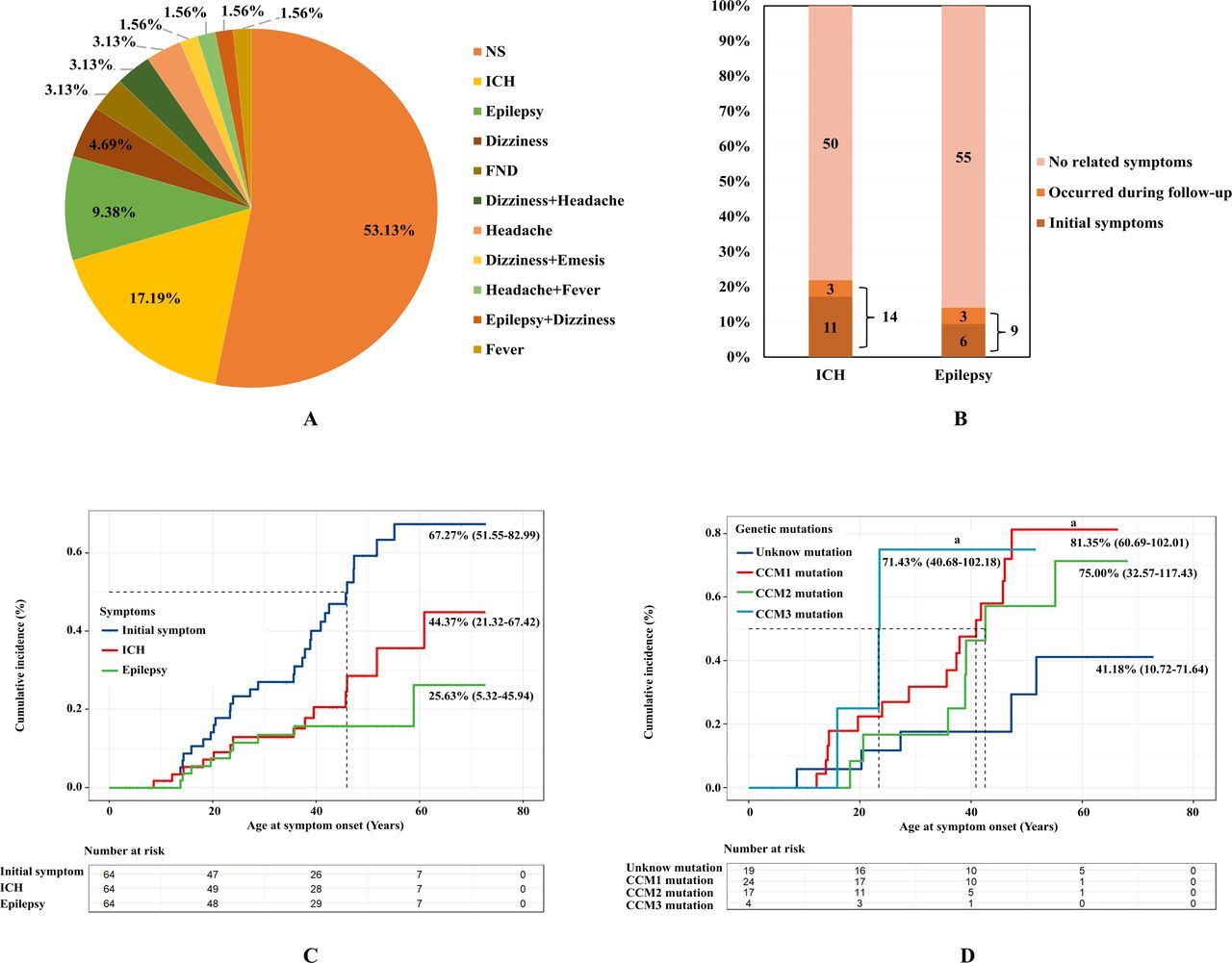
To determine the distribution of initial symptoms in FCCM, we investigated the initial symptoms experienced by 64 patients with FCCMs (figure 4A). In our cohort, 46.9% (n=30) were diagnosed with CCM after an initial presentation of neurological symptoms, indicating onset of disease. The three most common symptoms were ICH (n=11; 17.19%), epilepsy (n=6; 9.38%) and dizziness (n=3; 4.69%) (table 3). Using the KM method with initial symptoms as the outcome and age of onset as the follow time, results showed that the cumulative incidence of FCCM without intervention was 67.27% (51.55%–82.99%), and the mean age of symptom onset was 46.67 (40.56–52.78) years (figure 4C). Further analysis with bleeding and epilepsy as outcomes was performed to investigate the cumulative ICH and epilepsy rates in our cohort. To date, 14 patients with FCCMs developed ICH, whereas 9 patients developed epilepsy (figure 4B). Accordingly, the cumulative ICH rate was 44.37% (21.32%–67.42%), with a mean age of 58.19 (51.87–64.52) years (figure 4C), while the cumulative epilepsy rate was 25.63% (5.32%–45.94%, figure 4C). Lastly, KM analysis was used to investigate the cumulative incidence of FCCM in patients with different genotypes (table 3, figure 4D and online supplemental figure 2). Notably, patients with CCM1 (PBH=0.013) and CCM3 mutations (PBH=0.033) had a higher incidence of symptoms than those with unknown mutations. For ICH, the cumulative incidence in patients with CCM3 mutations was higher than in patients with unknown mutations (PBH=0.034). For epilepsy, the cumulative incidence was higher in patients with CCM3 mutations than in patients with unknown mutations (PBH=0.003) and CCM2 mutations (p=0.020). Similarly, higher cumulative epilepsy rates were observed in patients with CCM1 mutations compared with those with unknown mutations (p=0.038).
Incidence of different symptoms in patients with FCCMs (n=64)

Initial symptoms in patients with FCCMs (n=64). (A) Distribution of initial symptoms in patients with FCCMs. (B) Occurrence of ICH and epilepsy in patients with FCCMs. (C) Cumulative incidence of symptoms in patients in FCCMs. Blue lines represent the cumulative incidence of initial symptoms, red lines represent the cumulative incidence of ICH and green lines represent the cumulative incidence of seizures. (D) Cumulative incidence of FCCM patients with different gene mutations. FCCMs, familial cerebral cavernous malformations; ICH, intracranial haemorrhage.
Discussion
FCCM is a rare disorder characterised by clusters of mulberry-like or raspberry-like vascular malformations in the nervous system and is associated with multiple lesions.2 13 Currently, FCCM diagnosis is predominantly reliant on genetic testing and family medical history. In our clinical practice, we observed a higher number of lesions in patients with FCCMs than in those with multiple sporadic CCM lesions. Our study found that the presence of more than three CCM lesions on T2WI should serve as a strong indicator of FCCM, with a sensitivity and specificity of 87.10% and 87.50%, respectively. Our study compared sporadic probands with multiple CCMs among FCCM probands, which is relevant since most probands are individuals who are likely to seek medical attention. Previous studies have reported a greater number of lesions and larger average lesion size in patients with FCCMs compared with sporadic patients.14 15 The average total lesion count in patients with FCCM who seek medical attention, including multicentre data from the Brain Vascular Malformation Consortium (BVMC), was 13.5.16 Additionally, concurrent adrenal calcification may be present in these patients.17 Therefore, patients with over three CCM lesions on T2WI and with findings of adrenal calcification should be recommended for genetic testing and screening for FDRs in clinical practice.
Our study showed that 76.74% of families with multiple CCMs were diagnosed to have FCCM. This prevalence in China aligns with previous reports from Europe and America, where FCCM prevalence ranges from 80% to 90%.18 Since FCCM is an autosomal dominant genetic disorder, each affected parent has a 50% chance of transmitting the mutation to their offspring.2 However, few studies have reported the prevalence of FDRs in FCCM. In our study, FDRs of FCCM families were screened using cranial MRI and WES, revealing a prevalence of 36.5% (26.2%–46.7%). This high heritability, in addition to the significant burden FCCM places on the family, underscores the importance of screening. Therefore, the study provides evidence based to improve FCCM FDRs screening and policy development for the management and treatment. Comprehensive strategies such as preimplantation genetic testing and fetal MRI can be applied to reduce the prevalence of FCCM and prevent the disease from being inherited by offspring in the future.19 20
Previous studies investigating the distribution of known genes in FCCM have demonstrated significant variability. In our study, WES confirmed that 76.76% of families with FCCMs were found to have known CCM gene variant. Among these 25 families, the mutation rates of CCM1, CCM2 and CCM3 were 60.00% (15/25), 28.00% (7/25) and 12.00% (3/25), respectively. These findings are consistent with most previous studies in French, German and South Korean populations,8 13 21 22 where CCM1 accounts for 60%–70% of their cases, while CCM2 and CCM3 account for 10%–20% each. However, in the Italian population, CCM2 and CCM3 rates were, respectively, below 10%.23 Surprisingly, in the Japanese population, CCM1 accounted for 27.3% (3/11), CCM2 for 54.6% (6/11) and CCM3 for 18.2% (2/11) of their cohort.24 In comparison to other countries, large-sample surveys of FCCM gene distribution in Chinese populations are scarce. In one of these surveys involving 19 patients with multiple CCMs in Han Chinese of Taiwan, the mutation rates of CCM1, CCM2 and CCM3 were 80.0%, 6.7% and 13.3%, respectively.25 Discrepancies in mutation rate could be primarily attributed to the limited sample size. Overall, CCM1 expression was the highest in most populations, with variations in the distribution in different populations. On the other hand, CCM3 expression is the lowest among the three known genes but presents with more severe or earlier clinical presentations and imaging findings.26–28 A study by Scimone et al suggested that the asymmetric bidirectional promotion of CCM3 and SERPINI1 genes might play a protective role.29 At present, the HGMD database reports the identification of more than 320 mutations in CCM1, 110 mutations in CCM2 and 80 mutations in CCM3. However, several of these mutations remain unknown. Our study identified 10 novel mutations in known CCM genes, providing supporting evidence for the clinical genetic diagnoses. Additionally, eight FCCM families were found to have unidentified pathogenic genes, necessitating further exploration.
Unlike most previous studies that have investigated the distribution of symptoms among FCCM probands,2 30–32 this study aimed to capture a comprehensive picture of initial symptoms by including both probands and screened FDRs. Our study demonstrated that 53.13% of patients with FCCMs were asymptomatic, which is likely due to the inclusion of asymptomatic patients most from FDRs who were diagnosed by screening. This distribution of symptoms among patients with FCCMs diagnosed by screening closely resemble real-world prevalence. Asymptomatic patients may develop symptoms or lesions growth as age.13 16 27 Recent studies indicate that some medications can control the progression and reduce the burden of lesions in FCCM.33–36 Thus, asymptomatic patients may benefit from FCCM drug therapy in the near future. Our study also identified ICH as the most common symptom in symptomatic patients with FCCMs. This contradicts previous studies that reported epilepsy or headache as the most commonly reported symptom.2 14 16 37 38 Although, few studies support our finding, reporting ICH as the most common symptom.30 We believe that there are two main reasons for this discrepancy. First, ICH was the predominant symptom exhibited by patients in China. Second, our study classified neurological symptoms (eg, epilepsy, headache) secondary to ICH as bleeding events. Furthermore, cumulative incidence analysis of age at symptom onset revealed a mean age of 46.67 years for all symptoms and 58.19 years for initial ICH. These results provide valuable data for patient communication and FCCM management.
Despite the insights covered by this study, several limitations should be acknowledged. First, the FCCM families were enrolled at a single centre. Nevertheless, the neurosurgery department at our centre is the largest neurosurgery centre in Fujian Province, China. Therefore, most patients with multiple CCMs in the province visited our centre, and all enrolled families were from the Fujian Province. Second, the small family size in our study reflected the inherent rarity of FCCM as a disease condition. However, our study compensates for this by obtaining a large sample size through screening of FDRs, allowing a crude estimation of the prevalence of FDRs in FCCM. Lastly, some proband symptoms were observed retrospectively. A combination of face-to-face interviews by trained neurosurgeons or epidemiologists and standardised recruitment processes and procedures helped minimise recall bias.
Conclusion
This study investigated CCM in the FDRs of families with FCCMs were using MRI and genetic studies, resulting in a prevalence of 36.5% in China. Genetic counselling and MRI screening are recommended for FDRs in patients with more than three CCM lesions on T2WI in clinical practice. Further studies should also investigate the unidentified pathogenic genes that were found in approximately 24.24% of FCCM families.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the First Affiliated Hospital of Fujian Medical University Institutional Review Board (approval number: (2018) No. 003). Participants gave informed consent to participate in the study before taking part.
Footnotes
CL, LZ and YK contributed equally.
Contributors Concept and design: FL, DK, YL and SW. Participants contact and communication: CL, QL and DC. Information collection: CL, LZ, PL, QL, WZ, HW, QH and ZG. MRI scan, analysis: YK and WH. Drafting of the manuscript: CL and LZ. Statistical analysis: LZ and SL. Administrative, technical or material support: FL, LY, DW, BG, XN and YJ. Supervision: KM, SH and XL. Guarantor: FL.
Funding This work was supported by grants from Technology Platform Construction Project of Fujian Province (2020Y2003, 2021Y2001). And this work was supported the Fujian Province High level Neuromedical Center Construction Fund (principal investigator: DK), a grant from the Government of Fujian Province (grant number: HLNCC-FJFY-003). The word was also supported by grants from National Natural Science Foundation of China (8227051360).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
References




{kind=link}
{kind=link}
{kind=link}
{kind=link}