Multicentre prospective, randomised open-label, endpoint-blinded study to evaluate the safety and efficacy of propranolol for the prevention of stroke-associated pneumonia in patients with intracerebral haemorrhage (PROCHASE): rationale and design
- Bin Gao1,2,
- Kaibin Shi1,2,
- Yuesong Pan1,2,
- Shunnan Ge3,
- Yanfang Liu1,2,
- Jing Yan1,2,
- Arthur Liesz4,5,
- Andreas Meisel6,
- Yan Qu3,
- Xingquan Zhao1,2,
- Fu-Dong Shi1,2,7
- 1Department of Neurology, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
- 2China National Clinical Research Center for Neurological Diseases, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
- 3Department of Neurosurgery, Tangdu Hospital Fourth Military Medical University, Xi'an, China
- 4Institute for Stroke and Dementia Research (ISD), University Hospital, LMU Munich, Munich, Germany
- 5Munich Cluster for Systems Neurology (SyNergy), Munich, Germany
- 6Department of Neurology with Experimental Neurology, Neuroscience Clinical Research Center, Charité Universitätsmedizin Berlin, Berlin, Germany
- 7Department of Neurology, Tianjin Medical University General Hospital, Tianjin, China
- Correspondence to Dr Fu-Dong Shi; fshi{at}tmu.edu.cn; Professor Xingquan Zhao; zxq{at}vip.163.com
- Received 16 August 2024
- Accepted 17 December 2024
Abstract
Background Stroke-induced transient immune suppression is believed to contribute to post-stroke infections. The β-adrenergic receptor antagonist, propranolol, has been shown to prevent stroke-associated pneumonia (SAP) via reversing post-stroke immunosuppression in preclinical studies and in retrospective analysis in stroke patients. However, whether propranolol can reduce the risk of SAP has not been tested in prospective, randomised controlled trials.
Aim To describe the rationale and design of a multicentre, prospective, open-label, endpoint-blinded, randomised controlled study to evaluate the safety and efficacy of propranolol hydrochloride injection for the prevention of SAP in patients with intracerebral haemorrhage (ICH) (PROCHASE).
Design In this investigator-initiated trial, we compare the safety of the standard medical treatment to standard medical treatment plus intravenous propranolol hydrochloride administration (5 mg daily on days 1–7) in patients with ICH and the efficacy of this intervention to reduce the occurrence of SAP. All patients will be followed up for 90±7 days.
Study outcomes The primary efficacy outcome is SAP within 7±1 days diagnosed by the defined algorithm based on a diagnosis of SAP recommendations from the pneumonia in stroke consensus group. The primary safety outcome is defined as severe or moderate bradycardia within 7±1 days. The secondary outcome is a modified Rankin score of 0–3 at 90±7 days after randomisation.
Discussion The PROCHASE trial aims to generate clinical evidence regarding the safety and efficacy of propranolol in preventing SAP in patients with ICH.
- Stroke
- Infections
- Hemorrhage
WHAT IS ALREADY KNOWN ON THIS TOPIC
Stroke-associated pneumonia (SAP) significantly increases mortality in patients with intracerebral haemorrhage (ICH), and its pathogenesis is related to post-stroke immunosuppression. Propranolol has been shown to reverse post-stroke immune suppression in preclinical studies, but its safety and efficacy in preventing SAP in patients with ICH remain unclear.
WHAT THIS STUDY ADDS
The PROCHASE trial will deliver clinical evidence on the safety and efficacy of propranolol in lowering the risk of SAP following ICH.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The study will answer a key question of whether prevention of immunosuppression could prevent SAP and propose a novel and effective strategy for reducing the risk of this devastating complication following ICH.
Introduction and rationale
Approximately 30% of patients with stroke have infectious complications; among them, stroke-associated pneumonia (SAP) is the most prominent and severe one, occurring in approximately 10% of patients.1 2 Approximately half of SAP cases occur within 48 hours of stroke onset.3 SAP is associated with a higher risk of mortality and worse outcomes in stroke patients. The mortality rate in those with SAP is three times greater than in those without.4 Despite mixed results in several early phase studies,5 two large clinical trials demonstrated that preventive antibiotic use does not prevent SAP nor improve prognosis.6–8
Accumulating experimental and clinical evidence shows that the aetiology of SAP is related to stroke-induced immunosuppression, manifested as atrophy of immune organs, including the spleen, and contraction of lymphocyte populations.9 Mechanically, stroke activates the sympathetic nervous system (SNS), inducing lymphocyte apoptosis and lymphoid organ atrophy.9–12 Blockade of adrenergic signalling via the beta-blocker propranolol partially prevents the loss of lymphocytes and reverses post-stroke immunosuppression, reducing bacterial infection and death in animal models of stroke.10 13–15
Several retrospective studies have focused on the impact of beta-blockers on stroke prognosis; however, the results were mixed. A retrospective study involving 841 ischaemic stroke patients found that the use of beta-blockers after the onset of stroke was associated with a reduced 30 day mortality rate.16 However, in a single-centre prospective study of 1431 ischaemic stroke patients, beta-blocker use was associated with an increased incidence of infections.17 In a retrospective study of 138 hypertensive intracerebral haemorrhages (ICHs), the after-onset use of atenolol was associated with lower mortality within 30 days and reduced pneumonia.18 More recently, from a retrospective study involving 5212 stroke patients, after-onset beta-blockers reduced pneumonia within 10 days and mortality within 3 months.19 Therefore, the effectiveness of prophylactic beta-blockers in reducing the risk of SAP remains uncertain, highlighting the need for a prospective randomised controlled trial to address this issue.
Propranolol, a nonselective beta-blocker, can competitively block β1 and β2 receptors simultaneously and antagonise the effects of SNS excitation and catecholamine.20 Propranolol has long been approved for the treatment of hypertension, stable angina, tachycardic arrhythmia and other indications.21 22
Based on experimental and clinical evidence, we hypothesise that propranolol can reverse stroke-mediated immunosuppression and reduce the risk of SAP, thus improving stroke outcomes. In general, the prevalence of pneumonia after ICH is higher than ischaemic stroke (16.8% vs 11.7%).2 Furthermore, ICH has a higher incidence in China compared with developed countries (29·5% vs 15·8%)23 and higher mortality than ischaemic stroke.24 Therefore, we decided to focus on patients with ICH in this phase 2 study.
We plan to conduct a multicentre, prospective, open-label, endpoint-blind, randomised controlled, clinical trial to evaluate the safety and efficacy of propranolol in patients with ICH. We aim to test the hypothesis that blocking the SNS is capable of reversing immunosuppression and whether such a reversal could reduce the risk of post-stroke infections.
Methods
Design
The ongoing PROCHASE trial is an investigator-initiated, multicentre, prospective, randomised clinical trial with open-label treatment and blinded outcome assessments. The study is designed to recruit 168 patients over 2 years from comprehensive stroke centres across China. A flowchart drafting the study design is presented in figure 1. The study protocol underwent an amendment after the initial recruitment of 8% of patients. Both the original and amended protocols of PROCHASE have been approved by the ethics committees of all participating centres. Changes made to the original and amended protocols are detailed in the supplementary materials, and all enrolled patients met the criteria of the amended protocol.

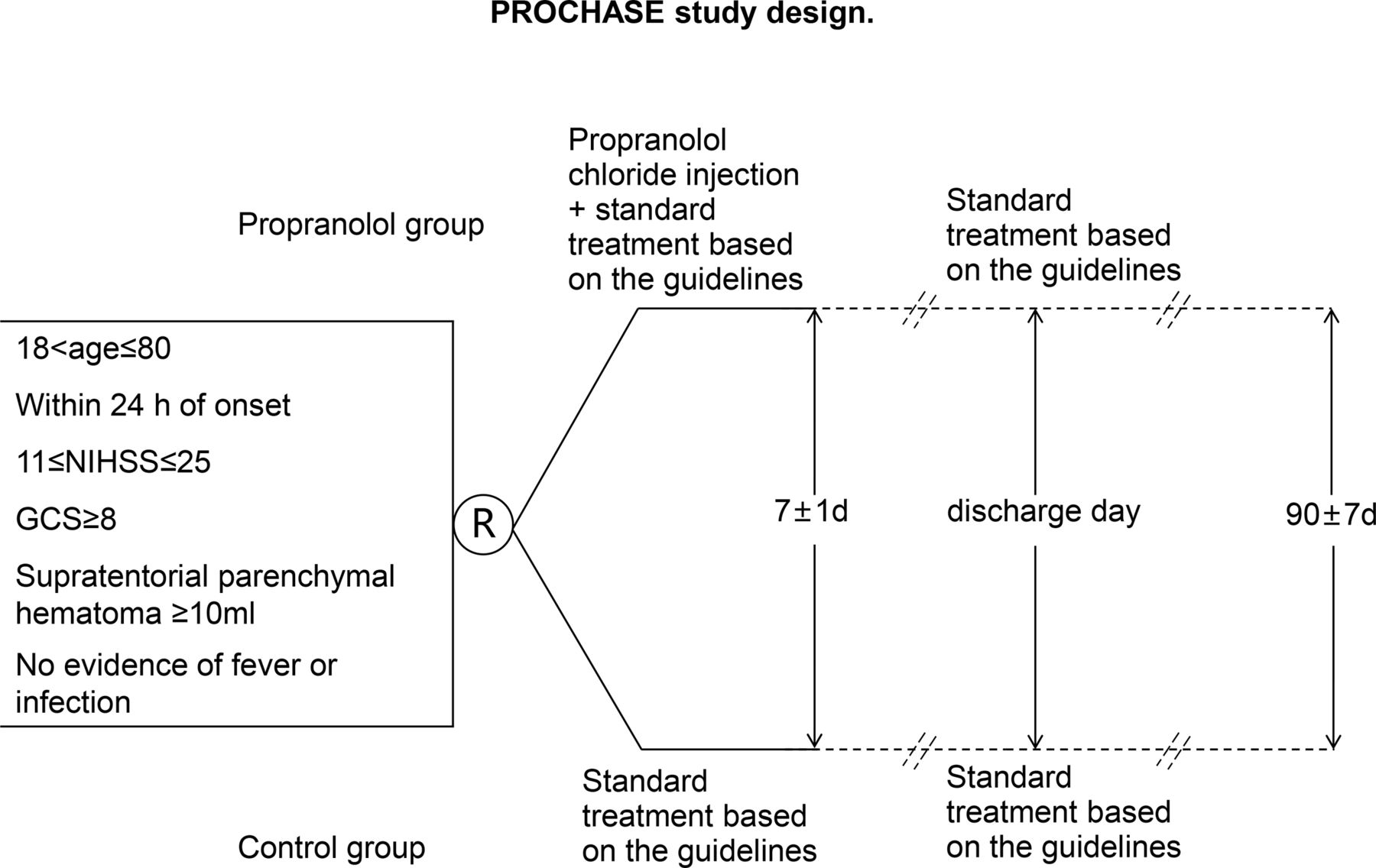{kind=link}
PROCHASE study design. PROCHASE, A multicentre prospective, randomised open-label, endpoint-blinded study to evaluate the safety and efficacy of propranolol for the prevention of SAP in patients with ICH.
Patient population
Participants are recruited from stroke centres across China. Patients with persistent neurological deficits who are diagnosed with ICH via head CT are screened in accordance with the study’s inclusion and exclusion criteria (online supplemental material). The inclusion criteria are as follows: patients aged >18 and ≤80, randomisation within 24 hours after ICH onset, no fever or evidence of infection on admission, National Institutes of Health Stroke Scale (NIHSS) score of 11–25, supratentorial parenchymal haematoma of ≥10 mL, Glasgow Coma Scale (GCS) of ≥8 points and patients or family members signed informed consent forms. Exclusion criteria are as follows: infections within the last 4 weeks, use of antibiotics within the last 2 weeks, known pre-ICH dysphagia, planned surgical treatment (including surgical removal of haematoma, decompressive craniectomy, minimally invasive haematoma aspiration and ventricular shunt or lateral ventricular drainage for ICH); primary ventricular haemorrhage or cerebral haemorrhage caused by trauma, vascular malformation, aneurysm, coagulopathy, anticoagulant or antiplatelet drugs, thrombolysis, post-infarction haemorrhagic transformation, haematopathy, moyamoya disease, primary or metastatic tumours, venous sinus thrombosis, vasculitis and other definite causes; history of stroke or pre-onset disability of limb motor function (modified Rankin scale (mRS) of ≥1); pregnancy or within 30 days of delivery; previous (within 1 month) use of beta-blockers or reserpine; bronchial asthma or chronic obstructive pulmonary disease; cardiogenic shock; II-III degree atrioventricular block; severe or acute heart failure; heart rate of <65 beats/min; known to be allergic to propranolol; severe liver or renal insufficiency; history of malignant tumour; currently participating in any other interventional clinical trials; and any treatment with immunosuppressants or immunotherapy.
Supplementary data
Randomisation
Following the obtaining of written informed consent, patients will be randomly assigned to the standard medical management group or standard medical management plus intravenous propranolol hydrochloride injection group in a 1:1 ratio. The randomisation process is internet-based and can be accessed via both mobile devices and web platforms. All participating centres will be subject to continuous monitoring to ensure adherence to the study protocol.
Treatment and intervention
Both treatment groups will receive standard medical care, which includes antihypertensive therapy, blood glucose management, intracranial pressure control or a combination of these interventions, in accordance with national and institutional guidelines. Patients are randomised within 24 hours after symptom onset or the timepoint of last seen well.
Intravenous propranolol hydrochloride injection (5 mg daily on day 1–7), provided by Jiangsu Longer Pharmaceutical Co, Ltd is initiated within 2 hour of randomisation if patients are allocated to the propranolol group. Detailed dosing guidelines were established for safety and presented in Supplementary materials.
Study organisation
On-site visits with participants are conducted separately at baseline, 72±12 hours, 7±1 days, 14±2 days or discharge day. Centralised telephone interviews are performed separately at 30±5 days and 90±7 days. NIHSS and GCS scores are recorded at baseline, 72±12 hours and 7±1 days. The mRS scores are recorded at baseline, 30±5 days and 90±7 days. Chest CT should be completed at baseline and conducted at any time within 7±1 days of onset if pneumonia is suspected by clinicians; otherwise, a chest CT will be conducted at 7±1 days. Head CT should be completed at baseline and 7±1 days. Laboratory tests and chest signs should be conducted at baseline, 72±12 hours and 7±1 days.
Primary outcome
SAP is diagnosed according to a defined algorithm within 7±1 days after randomisation. The predefined algorithm is based on the diagnosis of SAP recommendations from pneumonia in stroke consensus group (PISCES)25 and presented in online Supplementary materials.
Secondary outcomes
A favourable outcome is defined as an mRS of 0–3 on 90±7 days.
Safety outcomes
The primary safety outcome is defined as severe or moderate bradycardia within 7±1 days. Bradycardia is diagnosed using ECG monitoring. Severe bradycardia is defined as a heart rate of <40 beats/min. Moderate bradycardia is defined as a heart rate of <55 beats/min. Additional secondary safety outcomes include mortality, adverse events (AEs) and serious AEs, all occurring within 90 days, as reported by the investigators.
Study assessment
All patients undergo evaluation of the NIHSS and GCS at baseline, 72±12 hours and 7±1 days. At baseline, 72±12 hours and 7±1 day post-randomisation, body temperature was measured using an axillary thermometer, and pulmonary auscultation, laboratory tests and chest CT were performed to assess for SAP according to the algorithm defined by the diagnostic criteria from the PISCES group.25 Chest CT was repeated when patients exhibited SAP symptoms or 7 days post-randomisation. The 90 day clinical outcome, as defined by the mRS, was evaluated by independent investigators who were blinded to treatment allocation. All imaging data of the patients will be collected and independently evaluated by neurologists and radiologists, who were also blinded to treatment allocation. Video recordings of patients’ baseline neurological assessments are collected to allow an independent investigator to verify the adequate fulfilment of all the criteria of inclusion and exclusion prior to randomisation. Regular meetings with investigators are held to offer feedback and identify potential areas for improvement.
Sample size calculation
According to published retrospective studies,6 18 19 we predict the occurrence of SAP as follows:
30% of patients in the control group.6 26
9% of patients in the propranolol group, based on the efficacy data for propranolol in the prevention of SAP.8 19 In addition, the evaluated efficacy of atenolol in primary prevention reveals a relative 70% decrease in SAP risk in ICH patients.18 Based on these assumptions, and considering an alpha threshold of 5%, a power of 90% and an estimated risk of loss to follow-up of 20%, a minimum of 84 evaluable patients per group should be recruited, that is, a total of 168 patients. The sample size was calculated using PASS 21.0.2.
Statistical analyses
The primary outcome measure will be the risk ratio, which compares the proportion of patients who develop SAP within 7±1 days between the two treatment groups. This ratio will be adjusted for key prognostic factors such as age, baseline stroke severity (assessed by NIHSS and GCS), haematoma volume and other relevant variables that could contribute to imbalances between the intervention and control groups. Both intention-to-treat and per-protocol analyses will be conducted. Secondary outcomes will be analysed using log-binomial or linear regression methods, as appropriate, with the same adjustment approach applied as for the primary outcome. Missing baseline data will be reported, and any missing values will be imputed using regression-based methods. Adjusted and unadjusted estimates, along with the corresponding 95% CIs, will be presented. All analyses will be performed using a two-sided significance level of 5%.
Data safety and monitoring board
The data safety and monitoring board (DSMB) is made up of independent academic professionals and statisticians who are not involved in the trial’s implementation. The DSMB’s charter, outlining its membership, duties and responsibilities, will be reviewed and approved by both the board members and the executive committee. The DSMB will conduct continuous monitoring to ensure ethical standards and patient safety, with full access to the final dataset of the trial.
Management and quality control of data
Data will be transferred by an electronic data capture system using electronic case report forms (eCRFs), after being proofread and verified by data managers. All investigators at the participating sites are required to be qualified for good clinical practice (GCP). Clinical monitors will perform regular site visits to ensure full compliance with the study protocol and to verify that the eCRF records match the original data.
Withdrawal criteria for the study
Participants may withdraw from the study at any time, without the need to provide justification. If participants develop conditions that make them unsuitable for continued participation, the investigators will determine whether to withdraw them from the trial.
Management of project
The steering committee is in charge of designing the trial and providing overall strategic guidance. The executive committee oversees the study’s progress, with a particular focus on data collection. The adjudication committee, made up of external experts, will evaluate the study endpoints and AEs, making final decisions. Site investigators will manage patient recruitment, data collection as well as the reporting of clinical events. The National Clinical Research Centre for Neurological Diseases of China will manage the randomisation process, ensure data quality control and perform statistical analyses.
Discussion
The exaggerated activation of SNS following a stroke leads to systemic immunosuppression, characterised by lymphopenia and dysfunction of both peripheral innate and adaptive immune cells.10 13 These neuroimmune changes increase the risk of SAP and worsen patient outcomes after ICH. However, the preventive use of antibiotics fails to reduce the risk of SAP or improve the prognosis of stroke patients.8 27 From two decades ago, studies based on animal models have consistently demonstrated that propranolol could prevent stroke-induced immunosuppression and subsequent SAP; however, previous clinical observations generated conflicting results. Most previous clinical studies were retrospective and varied widely in several factors, including the patient population (primary vs recurrent stroke), treatment timing (pre-stroke with continuation post-stroke vs post-stroke only), stroke type (ischaemic vs haemorrhagic), β-blocker type (selective vs non-selective) and the definition of endpoints (variations in SAP assessment). Only one randomised controlled trial examined the use of propranolol in stroke patients, but SAP was not evaluated in the study.28 Furthermore, a standardised high dose of propranolol is challenging to implement in patients with ICH due to individual variability and the anticipated side effects. Therefore, we selected continuous intravenous infusion of propranolol with a predefined dosage, which can both avoid the absorption impairment of taking oral propranolol caused by gastrointestinal stress responses in patients with ICH and allow for the adjustment of pump speed to achieve the maximum dosage of patients with ICH under safe conditions.
In the past, there was a significant variation in the definition of SAP in different studies. Adhering to the latest guidelines for the diagnosis of SAP, SAP encompasses a range of lower respiratory tract infections occurring within the first 7 days post-ictus. The diagnosis of SAP is independent of pathogen culture and does not necessitate the differentiation between hospital-acquired pneumonia and community-acquired pneumonia. Chest imaging, particularly chest X-rays, is crucial in distinguishing between probable SAP and definite SAP. Further research has validated the high specificity and sensitivity of chest CT for the diagnosis of SAP.29
Continuous intravenous propranolol has a significant inhibitory effect on the heart rate. During administration, therefore, patients are continuously monitored via ECG during the treatment period of this study. If the heart rate is reduced below 55 beats per minute, an AE will be reported, and the attending physician will decide whether to continue the medication. If the heart rate is less than 40 beats per minute, propranolol will be immediately stopped, and the physician will determine the continuation of treatment based on the patient’s condition. Additionally, for patients experiencing tachycardia after medication of propranolol, the physician may increase the propranolol infusion rate to control the heart rate. Based on these considerations, the trial is designed as an open-label study, which is also one of the limitations of our trial. PROCHASE is the first randomised, multicentre, open-label, endpoint blinded clinical trial to determine the safety and efficacy of propranolol hydrochloride in preventing SAP in patients with moderate to severe ICH. If the study reveals positive trends, it could pave the way for a larger multicentre randomised clinical trial. Regardless of the therapeutic outcome, it will still provide important prospective data on the use of propranolol hydrochloride after ICH.
Trial status
Patient recruitment for this trial was still in progress at the time of manuscript submission. The first patient was enrolled on 21 February 2023. As of the submission of this manuscript, 138/168 patients have been recruited.
Summary and conclusions
The PROCHASE trial will produce reliable data on whether propranolol hydrochloride injection is capable of preventing SAP in patients with ICH.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. Data sharing is not applicable as no analyzable dataset was generated in this study.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by IRB of Beijing Tiantan Hospital, Capital Medical University, ID: KY2022-157-05. Participants gave informed consent to participate in the study before taking part.
Footnotes
Collaborators No available.
Contributors FDS and XZ had full access to all of the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: FDS, XZ and YQ. Draft of the manuscript: BG, KS, YP and SG. Critical revision of the manuscript for important intellectual content: AM and AL. Study supervision and organisation of the project: FDS, XZ, YQ, AM, AL, YL and JY. FDS is the guarantor.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.